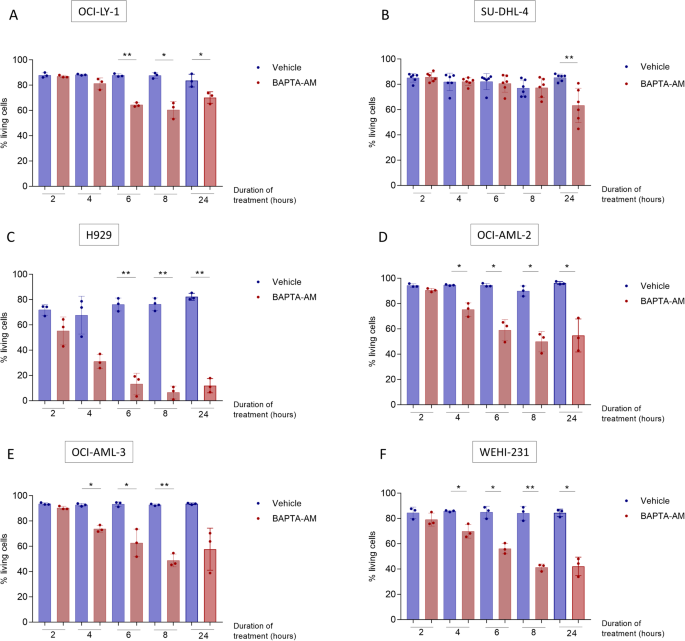
细胞内Ca 2+信号控制多种生理和病理生理过程。螯合细胞内Ca 2+的主要工具是细胞内BAPTA (BAPTA i ),通常以透膜乙酰氧基甲酯(BAPTA-AM) 的形式引入细胞中。此前,我们证明 BAPTA i可增强 BCL-2 拮抗剂 Venetoclax 在弥漫性大 B 细胞淋巴瘤 (DLBCL) 中诱导的细胞凋亡。这一发现暗示细胞内 Ca 2+信号传导和抗凋亡 BCL-2 功能之间存在新的相互作用。因此,我们着手确定 BAPTA i增强 B 细胞癌症细胞死亡的潜在机制。在这项研究中,我们发现 BAPTA i单独诱导对 MCL-1 拮抗剂 S63845 高度敏感的血液癌细胞系凋亡。BAPTA i通过抑制 mTORC1 驱动的Mcl-1翻译,引起 MCL-1 蛋白水平快速下降。这些事件并不是细胞死亡的结果,因为 BAX/BAK 缺陷的癌细胞表现出类似的 mTORC1 活性和 MCL-1 蛋白水平下调。接下来,我们研究了 BAPTA i如何降低 mTORC1 活性,并确定其通过直接抑制 6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶 3 (PFKFB3) 活性(BAPTA i 以前未知的作用)来损害糖酵解的能力。值得注意的是,这些效应也是由对 Ca 2+具有低亲和力的BAPTA i类似物引起的。因此,我们的研究结果揭示了 PFKFB3 抑制是一种独立于 Ca 2+的机制,BAPTA i通过该机制损害细胞代谢并最终损害 MCL-1 依赖性癌细胞的存活。这些发现有两个重要意义。首先,PFKFB3 的直接抑制成为 mTORC1 活性的关键调节因子,也是 MCL-1 依赖性癌症的一个有前景的靶点。其次,BAPTA i引起的细胞效应不一定与Ca 2+信号传导相关。当研究结果基于 BAPTA i的使用时,我们的数据支持重新评估 Ca 2+在细胞过程中的作用的必要性。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Intracellular BAPTA directly inhibits PFKFB3, thereby impeding mTORC1-driven Mcl-1 translation and killing MCL-1-addicted cancer cells
Intracellular Ca2+ signals control several physiological and pathophysiological processes. The main tool to chelate intracellular Ca2+ is intracellular BAPTA (BAPTAi), usually introduced into cells as a membrane-permeant acetoxymethyl ester (BAPTA-AM). Previously, we demonstrated that BAPTAi enhanced apoptosis induced by venetoclax, a BCL-2 antagonist, in diffuse large B-cell lymphoma (DLBCL). This finding implied a novel interplay between intracellular Ca2+ signaling and anti-apoptotic BCL-2 function. Hence, we set out to identify the underlying mechanisms by which BAPTAi enhances cell death in B-cell cancers. In this study, we discovered that BAPTAi alone induced apoptosis in hematological cancer cell lines that were highly sensitive to S63845, an MCL-1 antagonist. BAPTAi provoked a rapid decline in MCL-1-protein levels by inhibiting mTORC1-driven Mcl-1 translation. These events were not a consequence of cell death, as BAX/BAK-deficient cancer cells exhibited similar downregulation of mTORC1 activity and MCL-1-protein levels. Next, we investigated how BAPTAi diminished mTORC1 activity and identified its ability to impair glycolysis by directly inhibiting 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) activity, a previously unknown effect of BAPTAi. Notably, these effects were also induced by a BAPTAi analog with low affinity for Ca2+. Consequently, our findings uncover PFKFB3 inhibition as an Ca2+-independent mechanism through which BAPTAi impairs cellular metabolism and ultimately compromises the survival of MCL-1-dependent cancer cells. These findings hold two important implications. Firstly, the direct inhibition of PFKFB3 emerges as a key regulator of mTORC1 activity and a promising target in MCL-1-dependent cancers. Secondly, cellular effects caused by BAPTAi are not necessarily related to Ca2+ signaling. Our data support the need for a reassessment of the role of Ca2+ in cellular processes when findings were based on the use of BAPTAi.



































 京公网安备 11010802027423号
京公网安备 11010802027423号