当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate ab initio potential energy surface, rovibrational energy levels and resonance interactions of triplet (X~3B1) methylene
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2023-09-08 , DOI: 10.1002/jcc.27220 Oleg Egorov 1, 2 , Michaël Rey 3 , Dominika Viglaska 3 , Andrei V Nikitin 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2023-09-08 , DOI: 10.1002/jcc.27220 Oleg Egorov 1, 2 , Michaël Rey 3 , Dominika Viglaska 3 , Andrei V Nikitin 1
Affiliation
|
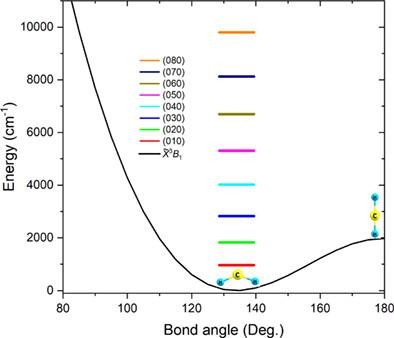
In this work, we report rovibrational energy levels for four isotopologues of methylene (CH2, CHD, CD2, and 13CH2) in their ground triplet electronic state (3B1) from variational calculation up to ~10,000 cm−1 and using a new accurate ab initio potential energy surface (PES). Triplet methylene exhibits a large-amplitude bending vibration and can reach a quasilinear configuration due to its low barrier (~2000 cm−1). To construct the ab initio PES, the Dunning's augmented correlation-consistent core-valence orbital basis sets were employed up to the sextuple-ζ quality [aug-cc-pCVXZ, X = T, Q, 5, and 6] combined with the single- and double-excitation unrestricted coupled cluster approach with a perturbative treatment of triple excitations [RHF-UCCSD(T)]. We have shown that the accuracy of the ab initio energies is further improved by including the corrections due to the scalar relativistic effects, DBOC and high-order electronic correlations. For the first time, all the available experimental rovibrational transitions were reproduced with errors less than 0.12 cm−1, without any empirical corrections. Unlike more “traditional” nonlinear triatomic molecules, we have shown that even the energies of the ground vibrational state (000) with rather small rotational quantum numbers are strongly affected by the very pronounced rovibrational resonance interactions. Accordingly, the polyad structure of the vibrational levels of CH2 and CD2 was analyzed and discussed. The comparison between the energy levels obtained from the effective Watson A-reduced Hamiltonian, from the generating-function approach and from a variational calculation was given.
中文翻译:

精确的从头算势能面、振动能级和三重态 (X~3B1) 亚甲基的共振相互作用
在这项工作中,我们报告了亚甲基的四种同位素体(CH 2、CHD、CD 2和13 CH 2)在其基态三重电子态下的振动能级(3 B 1)通过变分计算高达〜10,000 cm -1并使用新的精确从头势能表面(PES)。三重态亚甲基表现出大振幅的弯曲振动,并且由于其低势垒(~2000 cm -1)可以达到准线性构型。为了构建从头算 PES,邓宁的增强相关一致核价轨道基组被采用到六元- z 质量 [aug-cc-pCVXZ, X = T , Q, 5, 和 6] 与单- 以及对三重激发进行微扰处理的双激发无限制耦合簇方法 [RHF-UCCSD(T)]。我们已经证明,通过包含标量相对论效应、DBOC 和高阶电子相关性的修正,从头算能量的精度得到了进一步提高。第一次,所有可用的实验振动跃迁均以小于 0.12 cm -1的误差重现,无需任何经验修正。与更“传统”的非线性三原子分子不同,我们已经证明,即使具有相当小的旋转量子数的基振动态(000)的能量也会受到非常明显的振动共振相互作用的强烈影响。据此,对CH 2和CD 2振动能级的多聚体结构进行了分析和讨论。给出了从有效 Watson A简化哈密顿量、生成函数方法和变分计算获得的能级之间的比较。
更新日期:2023-09-08
中文翻译:
精确的从头算势能面、振动能级和三重态 (X~3B1) 亚甲基的共振相互作用
在这项工作中,我们报告了亚甲基的四种同位素体(CH 2、CHD、CD 2和13 CH 2)在其基态三重电子态下的振动能级(3 B 1)通过变分计算高达〜10,000 cm -1并使用新的精确从头势能表面(PES)。三重态亚甲基表现出大振幅的弯曲振动,并且由于其低势垒(~2000 cm -1)可以达到准线性构型。为了构建从头算 PES,邓宁的增强相关一致核价轨道基组被采用到六元- z 质量 [aug-cc-pCVXZ, X = T , Q, 5, 和 6] 与单- 以及对三重激发进行微扰处理的双激发无限制耦合簇方法 [RHF-UCCSD(T)]。我们已经证明,通过包含标量相对论效应、DBOC 和高阶电子相关性的修正,从头算能量的精度得到了进一步提高。第一次,所有可用的实验振动跃迁均以小于 0.12 cm -1的误差重现,无需任何经验修正。与更“传统”的非线性三原子分子不同,我们已经证明,即使具有相当小的旋转量子数的基振动态(000)的能量也会受到非常明显的振动共振相互作用的强烈影响。据此,对CH 2和CD 2振动能级的多聚体结构进行了分析和讨论。给出了从有效 Watson A简化哈密顿量、生成函数方法和变分计算获得的能级之间的比较。
































 京公网安备 11010802027423号
京公网安备 11010802027423号