当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Cryogenic Ion Vibrational Predissociation (CIVP) Spectroscopy of Aryl Cobinamides in the Gas Phase: How Good Are the Calculations for Vitamin B12 Derivatives?
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-09-01 , DOI: 10.1021/jacs.3c03001
Alexandra Tsybizova 1 , Lukas Fritsche 1 , Larisa Miloglyadova 1 , Bernhard Kräutler 2 , Peter Chen 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-09-01 , DOI: 10.1021/jacs.3c03001
Alexandra Tsybizova 1 , Lukas Fritsche 1 , Larisa Miloglyadova 1 , Bernhard Kräutler 2 , Peter Chen 1
Affiliation
|
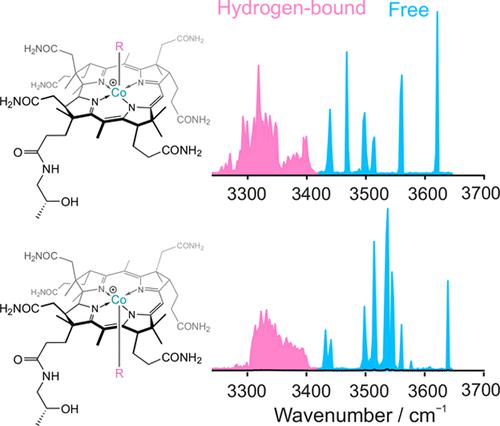
Aryl corrins represent a novel class of designed B12 derivatives with biological properties of “antivitamins B12”. In our previous study, we experimentally determined bond strength in a series of aryl-corrins by the threshold collision-induced dissociation experiments (T-CID) and compared the measured bond dissociation energies (BDEs) with those calculated with density functional theory (DFT). We found that the BDEs are modulated by the side chains around the periphery of the corrin unit. Given that aryl cobinamides have many side chains that increase their conformational space and that the question of a specific structure, measured in the gas phase, was important for further evaluation of our T-CID experiment, we proceeded to analyze structural properties of aryl cobinamides using cryogenic ion vibrational predissociation (CIVP) spectroscopy, static DFT, and Born–Oppenheimer molecular dynamic (BOMD) simulations. We found that none of the examined DFT models could reproduce the CIVP spectra convincingly; both “static” DFT calculations and “dynamic” BOMD simulations provide a surprisingly poor representation of the vibrational spectra, specifically of the number, position, and intensity of bands assigned to hydrogen-bonded versus non-hydrogen-bonded NH and OH moieties. We conclude that, for a flexible molecule with ca. 150 atoms, more accurate approaches are needed before definitive conclusions about computed properties, specifically the structure of the ground-state conformer, may be made.
中文翻译:

气相芳基钴酰胺的低温离子振动预解离 (CIVP) 光谱:维生素 B12 衍生物的计算效果如何?
芳基咕啉代表一类新型设计的 B 12衍生物,具有“抗维生素 B 12 ”的生物学特性。在我们之前的研究中,我们通过阈值碰撞诱导解离实验(T-CID)实验测定了一系列芳基咕啉的键强度,并将测得的键解离能(BDE)与密度泛函理论(DFT)计算的键解离能进行了比较。我们发现 BDE 是由 Corrin 单元周围的侧链调节的。鉴于芳基钴酰胺具有许多增加其构象空间的侧链,并且在气相中测量的特定结构问题对于进一步评估我们的 T-CID 实验非常重要,我们继续使用低温离子振动预解离 (CIVP) 光谱、静态 DFT 和玻恩-奥本海默分子动力学 (BOMD) 模拟。我们发现,所检查的 DFT 模型都无法令人信服地再现 CIVP 光谱; “静态”DFT 计算和“动态”BOMD 模拟都提供了令人惊讶的糟糕的振动光谱表示,特别是分配给氢键与非氢键 NH 和 OH 部分的带的数量、位置和强度。我们的结论是,对于具有约的柔性分子。 150 个原子,在得出有关计算特性(特别是基态构象异构体的结构)的明确结论之前,需要更准确的方法。
更新日期:2023-09-01
中文翻译:
气相芳基钴酰胺的低温离子振动预解离 (CIVP) 光谱:维生素 B12 衍生物的计算效果如何?
芳基咕啉代表一类新型设计的 B 12衍生物,具有“抗维生素 B 12 ”的生物学特性。在我们之前的研究中,我们通过阈值碰撞诱导解离实验(T-CID)实验测定了一系列芳基咕啉的键强度,并将测得的键解离能(BDE)与密度泛函理论(DFT)计算的键解离能进行了比较。我们发现 BDE 是由 Corrin 单元周围的侧链调节的。鉴于芳基钴酰胺具有许多增加其构象空间的侧链,并且在气相中测量的特定结构问题对于进一步评估我们的 T-CID 实验非常重要,我们继续使用低温离子振动预解离 (CIVP) 光谱、静态 DFT 和玻恩-奥本海默分子动力学 (BOMD) 模拟。我们发现,所检查的 DFT 模型都无法令人信服地再现 CIVP 光谱; “静态”DFT 计算和“动态”BOMD 模拟都提供了令人惊讶的糟糕的振动光谱表示,特别是分配给氢键与非氢键 NH 和 OH 部分的带的数量、位置和强度。我们的结论是,对于具有约的柔性分子。 150 个原子,在得出有关计算特性(特别是基态构象异构体的结构)的明确结论之前,需要更准确的方法。
































 京公网安备 11010802027423号
京公网安备 11010802027423号