当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Simulation of Metal–Ligand Self-Assembly into Spherical Complex M6L8
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2012-08-22 , DOI: 10.1021/ja303542r Makoto Yoneya 1 , Tomohiko Yamaguchi 1 , Sota Sato 2 , Makoto Fujita 2
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2012-08-22 , DOI: 10.1021/ja303542r Makoto Yoneya 1 , Tomohiko Yamaguchi 1 , Sota Sato 2 , Makoto Fujita 2
Affiliation
|
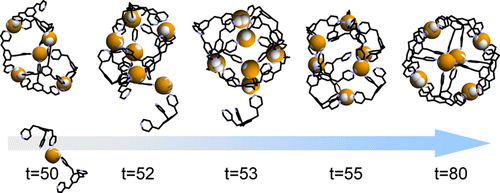
Molecular dynamics simulations were performed to study the self-assembly of a spherical complex through metal-ligand coordination interactions. M(6)L(8), a nanosphere with six palladium ions and eight pyridine-capped tridentate ligands, was selected as a target system. We successfully observed the spontaneous formation of spherical shaped M(6)L(8) cages over the course of our simulations, starting from random initial placement of the metals and ligands. To simulate spontaneous coordination bond formations and breaks, the cationic dummy atom method was employed to model nonbonded metal-ligand interactions. A coarse-grained solvent model was used to fill the gap between the time scale of the supramolecular self-assembly and that accessible by common molecular dynamics simulation. The simulated formation process occurred in the distinct three-stage (assembly, evolution, fixation) process that is well correlated with the experimental results. We found that the difference of the lifetime (or the ligand exchange rate) between the smaller-sized incomplete clusters and the completed M(6)L(8) nanospheres is crucially important in their supramolecular self-assembly.
中文翻译:

金属-配体自组装成球形复合物 M6L8 的模拟
进行分子动力学模拟以研究球形复合物通过金属-配体配位相互作用的自组装。M(6)L(8),具有六个钯离子和八个吡啶封顶的三齿配体的纳米球被选为目标系统。我们成功地观察到球形 M(6)L(8) 笼在我们的模拟过程中的自发形成,从金属和配体的随机初始放置开始。为了模拟自发配位键的形成和断裂,采用阳离子虚拟原子方法来模拟非键合金属-配体相互作用。粗粒度溶剂模型用于填补超分子自组装的时间尺度与常见分子动力学模拟的时间尺度之间的差距。模拟的形成过程发生在明显的三阶段(组装、演化、固定)过程中,与实验结果密切相关。我们发现较小尺寸的不完整簇和完整的 M(6)L(8) 纳米球之间的寿命(或配体交换率)的差异在其超分子自组装中至关重要。
更新日期:2012-08-22
中文翻译:
金属-配体自组装成球形复合物 M6L8 的模拟
进行分子动力学模拟以研究球形复合物通过金属-配体配位相互作用的自组装。M(6)L(8),具有六个钯离子和八个吡啶封顶的三齿配体的纳米球被选为目标系统。我们成功地观察到球形 M(6)L(8) 笼在我们的模拟过程中的自发形成,从金属和配体的随机初始放置开始。为了模拟自发配位键的形成和断裂,采用阳离子虚拟原子方法来模拟非键合金属-配体相互作用。粗粒度溶剂模型用于填补超分子自组装的时间尺度与常见分子动力学模拟的时间尺度之间的差距。模拟的形成过程发生在明显的三阶段(组装、演化、固定)过程中,与实验结果密切相关。我们发现较小尺寸的不完整簇和完整的 M(6)L(8) 纳米球之间的寿命(或配体交换率)的差异在其超分子自组装中至关重要。










































 京公网安备 11010802027423号
京公网安备 11010802027423号