Structure ( IF 4.4 ) Pub Date : 2023-08-31 , DOI: 10.1016/j.str.2023.08.008 Maximilian Kienlein 1 , Martin Zacharias 1 , Maria M Reif 1
|
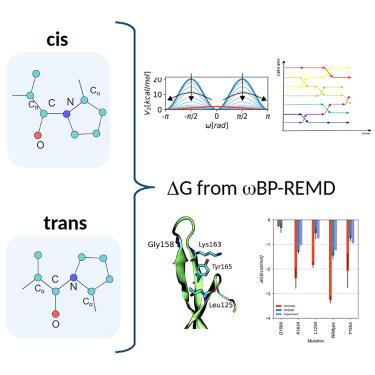
Proline cis/trans isomerization plays an important role in many biological processes but occurs on time scales not accessible to brute-force molecular dynamics (MD) simulations. We have designed a new Hamiltonian replica exchange scheme, ω-bias potential replica exchange molecular dynamics (ωBP-REMD), to efficiently and accurately calculate proline cis/trans isomerization free energies. ωBP-REMD is applied to various proline-containing tripeptides and a biologically important proline residue in the N2-domain of the gene-3-protein of phage fd in the wildtype and mutant variants of the protein. Excellent cis/trans transition rates are obtained. Reweighting of the sampled probability distribution along the peptide bond dihedral angle allows construction of the corresponding free-energy profile and calculation of the cis/trans isomerization free energy with high statistical precision. Very good agreement with experimental data is obtained. ωBP-REMD outperforms standard umbrella sampling in terms of convergence and agreement with experiment and strongly reduces perturbation of the local structure near the proline residue.
中文翻译:
通过哈密顿复制品交换分子动力学模拟高效准确地计算脯氨酸顺式/反式异构化自由能
脯氨酸顺式/反式异构化在许多生物过程中发挥着重要作用,但发生在强力分子动力学 (MD) 模拟无法达到的时间尺度上。我们设计了一种新的哈密顿复制品交换方案, ω-偏势复制品交换分子动力学( ω BP-REMD),以高效、准确地计算脯氨酸顺/反异构化自由能。 ω BP-REMD 适用于各种含脯氨酸的三肽以及噬菌体 fd 的野生型和突变型蛋白的基因 3 蛋白的 N2 结构域中生物学上重要的脯氨酸残基。获得优异的顺式/反式转变率。沿着肽键二面角重新加权采样的概率分布可以构建相应的自由能分布并以高统计精度计算顺式/反式异构化自由能。与实验数据非常吻合。 ω BP-REMD 在收敛性和与实验的一致性方面优于标准伞采样,并且大大减少了脯氨酸残基附近局部结构的扰动。






























 京公网安备 11010802027423号
京公网安备 11010802027423号