Bioorganic & Medicinal Chemistry Letters ( IF 2.5 ) Pub Date : 2023-08-29 , DOI: 10.1016/j.bmcl.2023.129462 Yanfei Zhang 1 , Jinlai Gao 2 , Jiming Wu 2 , Shihui Liu 2 , Xiaoping Zhang 3 , Xiaoqing Lv 1
|
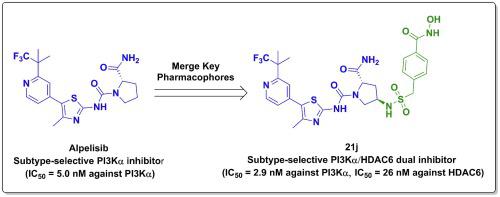
Recently, PI3K and HDAC have been considered as promising targets for the cancer therapy. A couple of pan-PI3K/HDAC dual inhibitors have been developed as a new class of anticancer agents. Herein, we discovered a new series of (S)-N1-(thiazol-2-yl) pyrrolidine-1,2-dicarboxamide derivatives targeting PI3Kα/HDAC6. All the derivatives exerted dual-target inhibitory activities. Particularly, in the enzymatic selectivity assay, compound 21j was identified as a subtype-selective PI3Kα/HDAC6 dual inhibitor (IC50 = 2.9 and 26 nM against PI3Kα and HDAC6, respectively), which displayed high potency against L-363 cell line with IC50 value of 0.17 μM. In addition, 21j significantly inhibited phosphorylation of pAkt(Ser473) and induced accumulation of acetylated α-tubulin while having a negligible effect on the levels of acetylated Histone H3 and H4 at nanomolar level. Attributed to its favorable in vitro performance, 21j has the potential to alleviate the adverse effects resulted from pan-PI3K inhibition and pan-HDAC inhibition. It is valuable for further functional investigation as an anti-cancer agent.
中文翻译:
发现靶向 PI3Ka/HDAC6 的 (S)-N1-(噻唑-2-基)吡咯烷-1,2-二甲酰胺衍生物用于治疗癌症
最近,PI3K和HDAC被认为是有希望的癌症治疗靶点。一些泛 PI3K/HDAC 双重抑制剂已被开发为一类新型抗癌药物。在此,我们发现了一系列针对 PI3Kα/HDAC6 的新 ( S )-N 1 - (thiazol-2-yl)pyrrolidine-1,2-dicarboxamide 衍生物。所有衍生物均发挥双靶点抑制活性。特别是,在酶选择性测定中,化合物21j被鉴定为亚型选择性 PI3Kα/HDAC6 双重抑制剂(针对 PI3Kα 和 HDAC6 的 IC 50 分别为 2.9 和 26 nM),对 L-363 细胞系显示出高效的 IC 值50值为 0.17 μM。此外,21j显着抑制 pAkt(Ser473) 的磷酸化并诱导乙酰化 α-微管蛋白的积累,同时对纳摩尔水平的乙酰化组蛋白 H3 和 H4 水平的影响可忽略不计。由于其良好的体外性能,21j有潜力减轻泛 PI3K 抑制和泛 HDAC 抑制引起的副作用。作为抗癌剂,它对于进一步的功能研究很有价值。





























 京公网安备 11010802027423号
京公网安备 11010802027423号