当前位置:
X-MOL 学术
›
Ind. Eng. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Atomic Connectivity Group Contribution Method for Predicting the Phase Transition Properties of Enthalpy and Entropy
Industrial & Engineering Chemistry Research ( IF 3.8 ) Pub Date : 2023-08-22 , DOI: 10.1021/acs.iecr.3c00971 Dongdong Cao 1 , Xiaojie Feng 1 , Qiang Wang 1 , Qingzhu Jia 2 , Shuqian Xia 3 , Fangyou Yan 1
Industrial & Engineering Chemistry Research ( IF 3.8 ) Pub Date : 2023-08-22 , DOI: 10.1021/acs.iecr.3c00971 Dongdong Cao 1 , Xiaojie Feng 1 , Qiang Wang 1 , Qingzhu Jia 2 , Shuqian Xia 3 , Fangyou Yan 1
Affiliation
|
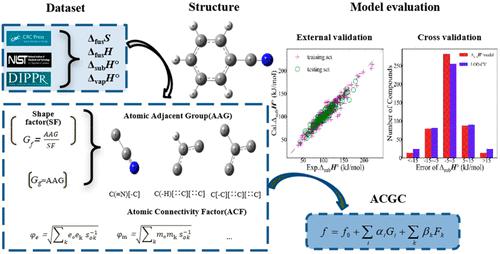
In this paper, four atomic connectivity group contribution (ACGC) models are developed for predicting the phase transition properties of organic compounds. These established ACGC models include atomic adjacent groups (AAG), shape factors (SFs), and atomic connectivity factors (ACFs), which are used to predict the entropy of fusion (ΔfusS), standard enthalpy of vaporization (ΔvapH°), standard enthalpy of sublimation (ΔsubH°), and enthalpy of fusion (ΔfusH). Groups are defined as atomic adjacent groups through the relation between the core atom and its neighbor atoms. Shape factors and atomic connectivity factors are proposed to deal with isomers in the group contribution (GC) method. After the introduction of SFs and ACFs into our models, the mean absolute error of the ΔfusS, ΔvapH°, ΔsubH°, and ΔfusH values for isomers decreases by 7.97%, 16.57%, 14.33%, and 6.28%, respectively. The ΔfusS, ΔvapH°, ΔsubH°, and ΔfusH models of ACGC can provide accurate calculation results with high R2 values of 0.945, 0.991, 0.921, and 0.901. Further, it is proved that ACGC models have good predictive ability and robustness through external validation and cross validation.
中文翻译:

预测焓和熵相变性质的原子连通基团贡献法
在本文中,开发了四种原子连接基团贡献(ACGC)模型来预测有机化合物的相变特性。这些建立的ACGC模型包括原子相邻组(AAG)、形状因子(SF)和原子连接因子(ACF),用于预测聚变熵(Δ fus S )、标准汽化焓(Δ vap H ° )、标准升华热 (Δ sub H° ) 和熔化热 (Δ fus H)。通过核心原子与其相邻原子之间的关系将基团定义为原子相邻基团。提出了形状因子和原子连接因子来处理基团贡献(GC)方法中的异构体。将 SF 和 ACF 引入模型后,异构体的 Δ fus S、Δ vap H°、Δ sub H°和 Δ fus H值的平均绝对误差分别降低了 7.97%、16.57%、14.33% 和分别为 6.28%。Δ fus S、 Δ vap H°、 Δ sub H°和 Δ fus HACGC模型可以提供精确的计算结果,R 2值高达0.945、0.991、0.921和0.901。进一步通过外部验证和交叉验证证明ACGC模型具有良好的预测能力和鲁棒性。
更新日期:2023-08-22
中文翻译:
预测焓和熵相变性质的原子连通基团贡献法
在本文中,开发了四种原子连接基团贡献(ACGC)模型来预测有机化合物的相变特性。这些建立的ACGC模型包括原子相邻组(AAG)、形状因子(SF)和原子连接因子(ACF),用于预测聚变熵(Δ fus S )、标准汽化焓(Δ vap H ° )、标准升华热 (Δ sub H° ) 和熔化热 (Δ fus H)。通过核心原子与其相邻原子之间的关系将基团定义为原子相邻基团。提出了形状因子和原子连接因子来处理基团贡献(GC)方法中的异构体。将 SF 和 ACF 引入模型后,异构体的 Δ fus S、Δ vap H°、Δ sub H°和 Δ fus H值的平均绝对误差分别降低了 7.97%、16.57%、14.33% 和分别为 6.28%。Δ fus S、 Δ vap H°、 Δ sub H°和 Δ fus HACGC模型可以提供精确的计算结果,R 2值高达0.945、0.991、0.921和0.901。进一步通过外部验证和交叉验证证明ACGC模型具有良好的预测能力和鲁棒性。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号