Acta Pharmaceutica Sinica B ( IF 14.7 ) Pub Date : 2023-08-17 , DOI: 10.1016/j.apsb.2023.08.015
Xuesong Li 1 , Xiang Chen 1 , Longbin Zheng 1, 2 , Minghong Chen 1 , Yunjia Zhang 1 , Ruigong Zhu 1 , Jiajing Chen 3 , Jiaming Gu 1 , Quanwen Yin 1 , Hong Jiang 1 , Xuan Wu 1 , Xian Ji 1 , Xin Tang 1 , Mengdie Dong 1 , Qingguo Li 4 , Yuanqing Gao 1 , Hongshan Chen 1, 4, 5, 6
|
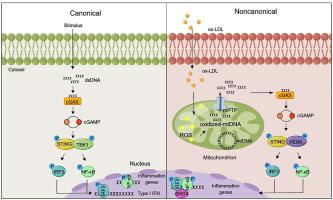
Inflammation-driven endothelial dysfunction is the major initiating factor in atherosclerosis, while the underlying mechanism remains elusive. Here, we report that the non-canonical stimulator of interferon genes (STING)–PKR-like ER kinase (PERK) pathway was significantly activated in both human and mice atherosclerotic arteries. Typically, STING activation leads to the activation of interferon regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB)/p65, thereby facilitating IFN signals and inflammation. In contrast, our study reveals the activated non-canonical STING–PERK pathway increases scaffold protein bromodomain protein 4 (BRD4) expression, which encourages the formation of super-enhancers on the proximal promoter regions of the proinflammatory cytokines, thereby enabling the transactivation of these cytokines by integrating activated IRF3 and NF-κB via a condensation process. Endothelium-specific STING and BRD4 deficiency significantly decreased the plaque area and inflammation. Mechanistically, this pathway is triggered by leaked mitochondrial DNA (mtDNA) via mitochondrial permeability transition pore (mPTP), formed by voltage-dependent anion channel 1 (VDAC1) oligomer interaction with oxidized mtDNA upon cholesterol oxidation stimulation. Especially, compared to macrophages, endothelial STING activation plays a more pronounced role in atherosclerosis. We propose a non-canonical STING–PERK pathway-dependent epigenetic paradigm in atherosclerosis that integrates IRF3, NF-κB and BRD4 in inflammatory responses, which provides emerging therapeutic modalities for vascular endothelial dysfunction.
中文翻译:
通过在炎症反应中整合 IRF3 和 NF-κB,对血管内皮功能障碍进行非经典 STING-PERK 通路依赖性表观遗传调控
炎症驱动的内皮功能障碍是动脉粥样硬化的主要引发因素,但其潜在机制仍不清楚。在这里,我们报告干扰素基因的非经典刺激物(STING)-PKR 样 ER 激酶(PERK)通路在人类和小鼠动脉粥样硬化动脉中显着激活。通常,STING 激活会导致干扰素调节因子 3 (IRF3) 和核因子 kappa B (NF- κ B)/p65 的激活,从而促进IFN信号和炎症。相比之下,我们的研究表明,激活的非经典 STING-PERK 通路会增加支架蛋白溴结构域蛋白 4 (BRD4) 的表达,从而促进促炎细胞因子的近端启动子区域形成超级增强子,从而使这些细胞因子的反式激活成为可能。通过缩合过程整合激活的 IRF3 和 NF- κ B 来抑制细胞因子。内皮特异性 STING 和 BRD4 缺陷显着减少了斑块面积和炎症。从机制上讲,该途径是由线粒体 DNA (mtDNA)通过线粒体通透性转换孔 (mPTP) 泄漏而触发的,mPTP 是由电压依赖性阴离子通道 1 (VDAC1) 寡聚物在胆固醇氧化刺激下与氧化 mtDNA 相互作用形成的。特别是,与巨噬细胞相比,内皮 STING 激活在动脉粥样硬化中发挥着更显着的作用。我们提出了一种非经典的动脉粥样硬化 STING-PERK 通路依赖性表观遗传范例,它将 IRF3、NF- κ B 和 BRD4 整合到炎症反应中,为血管内皮功能障碍提供了新兴的治疗方式。
































 京公网安备 11010802027423号
京公网安备 11010802027423号