Journal of Catalysis ( IF 6.5 ) Pub Date : 2023-08-09 , DOI: 10.1016/j.jcat.2023.08.009 Yi Feng , Ji Qi , Maoshuai Li , Jie Wei , Qi Yang , Jie Ding , Mei-Yan Wang , Xinbin Ma
|
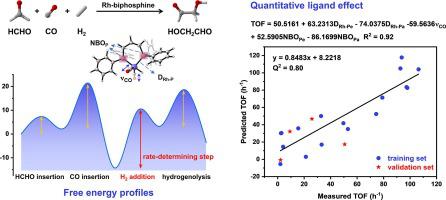
Hydroformylation is one of the most important homogenous reactions with more than 10 million tons of aldehydes production each year. Due to monodentate ligands instable coordination under hydroformylation of formaldehyde, here, we introduced a range of chelating biphosphine ligands onto rhodium catalysts. Ligands, such as BINAP (TOFGA = 106.4 h−1), present higher activity relative to common monodentate ligand PPh3 (TOFGA = 73.4 h−1) with lower ligand to metal ratio. Density functional theory calculation revealed that Rh-biphosphine complexes follow the anion mechanism, with H2 oxidation addition being the rate-determining step. Activation strain analysis further demonstrated that lower H2 distortion energy resulting higher TOF of glycolaldehyde formation. Besides, the predictable quantitative structure–activity relationship model was established and suggested that both electronic and steric properties synergistically controlled formaldehyde hydroformylation activity. This combination of mathematical modeling and theoretical calculation will provide a reliable approach to activity prediction and catalyst design for important industrial hydroformylation reaction.
中文翻译:
Rh-双膦配合物在甲醛加氢甲酰化反应中的定量配体效应及机理研究
加氢甲酰化是最重要的均相反应之一,每年醛类产量超过1000万吨。由于单齿配体在甲醛加氢甲酰化下配位不稳定,在这里,我们在铑催化剂上引入了一系列螯合双膦配体。配体,例如BINAP (TOF GA = 106.4 h -1 ),相对于配体与金属比率较低的常见单齿配体PPh 3 (TOF GA = 73.4 h -1 ) 表现出更高的活性。密度泛函理论计算表明Rh-双膦配合物遵循阴离子机理,其中H 2氧化加成是速率决定步骤。活化应变分析进一步表明,较低的H 2畸变能导致乙醇醛形成的TOF较高。此外,建立了可预测的定量结构-活性关系模型,并表明电子和空间性质协同控制甲醛加氢甲酰化活性。数学建模和理论计算的结合将为重要的工业加氢甲酰化反应的活性预测和催化剂设计提供可靠的方法。
































 京公网安备 11010802027423号
京公网安备 11010802027423号