当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crystalline Arrays of Copper Porphyrin Qubits Based on Ion-Paired Frameworks
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-08-08 , DOI: 10.1021/jacs.3c04786 Casandra M Moisanu 1 , Robert M Jacobberger 1 , Luke P Skala 1 , Charlotte L Stern 1 , Michael R Wasielewski 1 , William R Dichtel 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-08-08 , DOI: 10.1021/jacs.3c04786 Casandra M Moisanu 1 , Robert M Jacobberger 1 , Luke P Skala 1 , Charlotte L Stern 1 , Michael R Wasielewski 1 , William R Dichtel 1
Affiliation
|
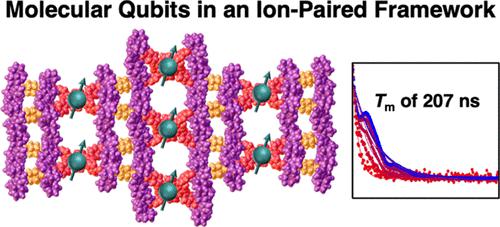
Molecular electronic spin qubits have great potential for use in quantum information science applications because their structure can be rationally tuned using synthetic chemistry. Their integration into a new class of materials, ion-paired frameworks, allows for the formation of ordered arrays of these molecular spin qubits. Three ion-paired frameworks with varying densities of paramagnetic Cu(II) porphyrins were isolated as micron-sized crystals suitable for characterization by single-crystal X-ray diffraction. Pulse-electron paramagnetic resonance (EPR) spectroscopy probed the spin coherence of these materials at temperatures up to 140 K. The crystals with the longest Cu–Cu distances had a spin–spin relaxation time (Tm) of 207 ns and a spin–lattice relaxation time (T1) of 1.8 ms at 5 K, which decreased at elevated temperature because of spin–phonon coupling. Crystals with shorter Cu–Cu distances also had lower T1 values because of enhanced cross-relaxation from qubit–qubit dipolar coupling. Frameworks with shorter Cu–Cu distances exhibited lower Tm values because of the increased interactions between qubits within the frameworks. Incorporating molecular electronic spin qubits in ion-paired frameworks enables control of composition, spacing, and interqubit interactions, providing a rational means to extend spin relaxation times.
中文翻译:

基于离子对骨架的铜卟啉量子位晶体阵列
分子电子自旋量子位在量子信息科学应用中具有巨大的潜力,因为它们的结构可以使用合成化学进行合理调整。它们集成到一类新型材料——离子对框架中,可以形成这些分子自旋量子位的有序阵列。三种具有不同密度顺磁性 Cu(II) 卟啉的离子对骨架被分离为微米级晶体,适合通过单晶 X 射线衍射表征。脉冲电子顺磁共振 (EPR) 光谱在高达 140 K 的温度下探测了这些材料的自旋相干性。具有最长 Cu-Cu 距离的晶体具有 207 ns 的自旋-自旋弛豫时间 (T m )和自旋- 5 K 时晶格弛豫时间 ( T 1 ) 为 1.8 ms,由于自旋声子耦合,晶格弛豫时间在升高的温度下会降低。由于量子位-量子位偶极耦合的交叉弛豫增强,Cu-Cu 距离较短的晶体也具有较低的T 1值。Cu-Cu 距离较短的框架表现出较低的T m值,因为框架内量子位之间的相互作用增加。将分子电子自旋量子位纳入离子对框架中,可以控制组成、间距和量子位间相互作用,从而提供延长自旋弛豫时间的合理方法。
更新日期:2023-08-08
中文翻译:
基于离子对骨架的铜卟啉量子位晶体阵列
分子电子自旋量子位在量子信息科学应用中具有巨大的潜力,因为它们的结构可以使用合成化学进行合理调整。它们集成到一类新型材料——离子对框架中,可以形成这些分子自旋量子位的有序阵列。三种具有不同密度顺磁性 Cu(II) 卟啉的离子对骨架被分离为微米级晶体,适合通过单晶 X 射线衍射表征。脉冲电子顺磁共振 (EPR) 光谱在高达 140 K 的温度下探测了这些材料的自旋相干性。具有最长 Cu-Cu 距离的晶体具有 207 ns 的自旋-自旋弛豫时间 (T m )和自旋- 5 K 时晶格弛豫时间 ( T 1 ) 为 1.8 ms,由于自旋声子耦合,晶格弛豫时间在升高的温度下会降低。由于量子位-量子位偶极耦合的交叉弛豫增强,Cu-Cu 距离较短的晶体也具有较低的T 1值。Cu-Cu 距离较短的框架表现出较低的T m值,因为框架内量子位之间的相互作用增加。将分子电子自旋量子位纳入离子对框架中,可以控制组成、间距和量子位间相互作用,从而提供延长自旋弛豫时间的合理方法。
































 京公网安备 11010802027423号
京公网安备 11010802027423号