Journal of Molecular Modeling ( IF 2.1 ) Pub Date : 2023-07-24 , DOI: 10.1007/s00894-023-05666-6 Christian Tshikala Mukeba 1, 2 , Bienfait Kabuyaya Isamura 1, 2, 3 , Virima Mudogo 1, 4 , Haddy Mbuyi Katshiatshia 5 , Jules Tshishimbi Muya 1, 2
|
Context
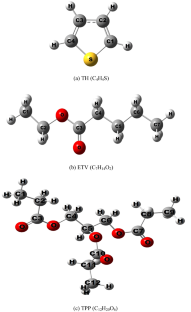
Due to the expected decrease in the availability of conventional oils, numerous studies are currently underway to find complementary sources of energy. Among the explored avenue is that of biofuels. Ethyl valerate (ETV) and tripropionin (TPP) are two biofuels whose thermal decomposition has not received the attention it deserves. Herein, we have evaluated the bond dissociation enthalpies (BDHs) to predict how easy it is to break some bonds in these compounds, and subsequently contribute to revealing the initiation step in their combustion reactions. Our computations consistently predict C4-C5 and C1-C2 bonds in ETV and TPP as the weakest bonds, likely to break first and initiate the thermal decomposition of these two compounds, respectively. The conformational changes in ETV and TPP have only a small influence on the BDHs of 1 kcal/mol at M06-2X/6–311 + G(3df,2p). B3LYP and ωB97XD appear to be the most affordable methods for estimating BDHs at 6-31G(d,p) as they give good results for ETV (RMSD: 2.94 kcal/mol and 3.22 kcal/mol) and performed better than CBS-QB3 (RMSD: 3.64 kcal/mol). Using a larger basis set, the M06-2X (RMSD: 3.61 kcal/mol) and ωB97XD (RMSD: 3.51 kcal/mol) functionals are found to provide the most accurate predictions at 6–311 + G(3df,2p) as compared to G4MP2.
Methods
BDHs of ETV and TPP are computed using density functional theory (DFT) and quantum chemistry composite methods at 6-31G(d,p) and 6–311 + G(3df,2p) levels. Because of its reliability and accuracy in thermochemical calculations, the G4MP2 theory is used as a reference to gauge the performance of DFT methods. All the calculations were carried out using the Gaussian 09 program.
中文翻译:
戊酸乙酯和三丙酸的键解离能
语境
由于传统石油的供应预计会减少,目前正在进行大量研究来寻找补充能源。已探索的途径之一是生物燃料。戊酸乙酯(ETV)和三丙酸甘油酯(TPP)是两种生物燃料,其热分解尚未受到应有的关注。在此,我们评估了键解离焓 (BDH),以预测破坏这些化合物中某些键的难易程度,从而有助于揭示其燃烧反应的引发步骤。我们的计算一致预测 ETV 和 TPP 中的 C4-C5 和 C1-C2 键是最弱的键,可能首先断裂并分别引发这两种化合物的热分解。ETV 和 TPP 的构象变化对 M06-2X/6–311 + G(3df,2p) 处 1 kcal/mol 的 BDH 仅产生很小的影响。B3LYP 和 ωB97XD 似乎是估计 6-31G(d,p) 处 BDH 的最经济实惠的方法,因为它们为 ETV 提供了良好的结果(RMSD:2.94 kcal/mol 和 3.22 kcal/mol)并且比 CBS-QB3 表现更好( RMSD:3.64 kcal/mol)。相比之下,使用更大的基组,M06-2X(RMSD:3.61 kcal/mol)和 ωB97XD(RMSD:3.51 kcal/mol)泛函可在 6–311 + G(3df,2p) 处提供最准确的预测到 G4MP2。
方法
ETV 和 TPP 的 BDH 使用密度泛函理论 (DFT) 和量子化学复合方法在 6-31G(d,p) 和 6-311 + G(3df,2p) 水平上计算。由于其在热化学计算中的可靠性和准确性,G4MP2 理论被用作衡量 DFT 方法性能的参考。所有计算均使用Gaussian 09程序进行。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号