Journal of Molecular and Cellular Cardiology ( IF 4.9 ) Pub Date : 2023-07-21 , DOI: 10.1016/j.yjmcc.2023.07.008
Yuanyuan Chen 1 , Yuchan Yuan 1 , Yuhan Chen 1 , Xueze Jiang 1 , Xuesheng Hua 1 , Zhiyong Chen 1 , Julie Wang 2 , Hua Liu 3 , Qing Zhou 1 , Ying Yu 1 , Zhenwei Yang 1 , Yi Yu 1 , Yongqin Wang 4 , Qunshan Wang 1 , Yigang Li 1 , Jie Chen 1 , Yuepeng Wang 1
|
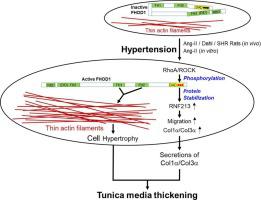
Hypertension-induced tunica media thickening (TMT) is the most important fundamental for the subsequent complications like stroke and cardiovascular diseases. Pathogenically, TMT originates from both vascular smooth muscle cells (VSMCs) hypertrophy due to synthesizing more amount of intracellular contractile proteins and excess secretion of extracellular matrix. However, what key molecules are involved in the pathogenesis of TMT is unknown. We hypothesize that formin homology 2 domain-containing protein 1 (FHOD1), an amply expressed mediator for assembly of thin actin filament in VSMCs, is a key regulator for the pathogenesis of TMT. In this study, we found that FHOD1 expression and its phosphorylation/activation were both upregulated in the arteries of three kinds of hypertensive rats. Ang-II induced actin filament formation and hypertrophy through activation and upregulation of FHOD1 in VSMCs. Active FHOD1-mediated actin filament assembly and secretions of collagen-1α/collagen-3α played crucial roles in Ang-II-induced VSMCs hypertrophy in vitro and hypertensive TMT in vivo. Proteomics demonstrated that activated FL-FHOD1 or its C-terminal diaphanous-autoregulatory domain significantly upregulated RNF213 (ring finger protein 213), a 591-kDa cytosolic E3 ubiquitin ligase with its loss-of-functional mutations being a susceptibility gene for Moyamoya disease which has prominent tunica media thinning in both intracranial and systemic arteries. Mechanistically, activated FHOD1 upregulated its downstream effector RNF213 independently of its classical pathway of decreasing G-actin/F-actin ratio, transcription, and translation, but dependently on its C-terminus-mediated stabilization of RNF213 protein. FHOD1-RNF213 signaling dramatically promoted collagen-1α/collagen-3α syntheses in VSMCs. Our results discovered a novel signaling axis of FHOD1-RNF213-collagen-1α/collagen-3α and its key role in the pathogenesis of hypertensive TMT.
中文翻译:
FHOD1-RNF213-Col1α/Col3α在高血压引起的中膜增厚发病机制中的新信号轴
高血压引起的中膜增厚(TMT)是中风、心血管疾病等后续并发症的最重要基础。从病理上看,TMT源于血管平滑肌细胞(VSMCs)因细胞内收缩蛋白合成过多和细胞外基质过度分泌而肥大。然而,哪些关键分子参与TMT的发病机制尚不清楚。我们假设含有福尔马林同源性 2 结构域的蛋白 1 (FHOD1) 是 VSMC 中细肌动蛋白丝组装的充分表达介导物,是 TMT 发病机制的关键调节因子。在本研究中,我们发现三种高血压大鼠的动脉中FHOD1表达及其磷酸化/激活均上调。 Ang-II 通过激活和上调 VSMC 中的 FHOD1 诱导肌动蛋白丝形成和肥大。活性 FHOD1 介导的肌动蛋白丝组装和胶原蛋白 1α/胶原蛋白 3α 的分泌在 Ang-II 诱导的体外VSMC 肥大和体内高血压 TMT 中发挥着至关重要的作用。蛋白质组学证明,激活的 FL-FHOD1 或其 C 端透明自动调节结构域显着上调 RNF213(环指蛋白 213),RNF213 是一种 591 kDa 胞质 E3 泛素连接酶,其功能丧失突变是烟雾病的易感基因,颅内动脉和全身动脉中膜均显着变薄。从机制上讲,激活的 FHOD1 上调其下游效应子 RNF213,独立于其降低 G-肌动蛋白/F-肌动蛋白比率、转录和翻译的经典途径,但依赖于其 C 端介导的 RNF213 蛋白的稳定性。 FHOD1-RNF213 信号传导显着促进 VSMC 中胶原蛋白 1α/胶原蛋白 3α 的合成。我们的研究结果发现了 FHOD1-RNF213-collagen-1α/collagen-3α 的新信号轴及其在高血压 TMT 发病机制中的关键作用。
































 京公网安备 11010802027423号
京公网安备 11010802027423号