Chemical Physics ( IF 2.0 ) Pub Date : 2023-07-18 , DOI: 10.1016/j.chemphys.2023.112018
Shehu Aminu Yamusa , Amiruddin Shaari , Norah A.M. Alsaif , Najeh Rekik , G. Lakshminarayana , Ibrahim Isah , Magaji Ismail , Razif Razali
|
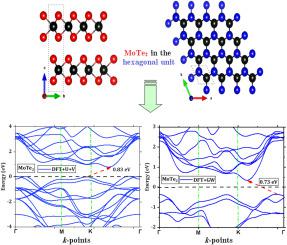
Molybdenum ditelluride (MoTe) is a promising two-dimensional material with ultimate prospective usage in high performance photodetection devices. In this study, we elucidate how this may be revealed and discuss how structural and optoelectronic properties of MoTe can be numerically accurately simulated since earlier experimental and theoretical studies on the bandgap of MoTe produced contradictory findings. In doing so, GW-based functionals using Hubbard U and V corrections are included in density functional theory (DFT) calculations to improve bandgap estimations. Interestingly, we reliably demonstrated that the estimated values of the bandgaps of 0.83 eV and 0.73 eV obtained, respectively, within this framework of DFT+U+V and GW, perfectly match the reported experimental results. Specifically, the quantum espresso simulation package is used for accurate DFT calculations allowing thereby a comprehensive investigation of the impact of the Hubbard U correction on the bandgap of MoTe. Additionally, the optical absorption spectrum is examined for both GW and RPA levels of theory using the Yambo simulation tool, allowing for a readily distinctly identification of the material’s light absorption spectrum. Contrasted by previous theoretical results, the random phase approximation (RPA) approach, which performs quite well in showing increased optical efficiency, reveals its effectiveness for obtaining appreciable gains in the values of the real, imaginary, refractive index, and extinction coefficient. The expected trends obtained with GW-based functionals using Hubbard U and V corrections approximate methods are encouraging, and altogether support ongoing attempts to optimize the physical properties of MoTe for high-performance photodetection systems by offering more precise bandgap predictions and valuable insights related particularly to the optical properties.
中文翻译:
用于高性能光电探测的 MoTe2 结构和光电特性的增强 DFT 预测:应用于基于 GW 的泛函和 Hubbard U 和 V 校正
二碲化钼 (MoTe)是一种有前途的二维材料,在高性能光电检测设备中具有最终的应用前景。在这项研究中,我们阐明了如何揭示这一点,并讨论了 MoTe 的结构和光电特性如何自从早期对 MoTe 带隙的实验和理论研究以来,可以进行精确的数值模拟产生了矛盾的发现。在此过程中,使用 Hubbard U 和 V 校正的基于 GW 的泛函被包含在密度泛函理论 (DFT) 计算中,以改进带隙估计。有趣的是,我们可靠地证明了在 DFT+U+V 和 GW 的框架内分别获得的带隙估计值 0.83 eV 和 0.73 eV 与报告的实验结果完全匹配。具体来说,量子浓缩咖啡模拟包用于精确的 DFT 计算,从而可以全面研究 Hubbard U 校正对 MoTe 带隙的影响。此外,使用 Yambo 模拟工具检查光吸收光谱的GW 和 RPA理论水平,从而可以轻松清晰地识别材料的光吸收光谱。与之前的理论结果相比,随机相位近似(RPA)方法在提高光学效率方面表现良好,揭示了其在实部、虚部、折射率和消光系数值方面获得可观增益的有效性。使用 Hubbard U 和 V 校正近似方法基于 GW 泛函获得的预期趋势令人鼓舞,并且完全支持正在进行的优化 MoTe 物理特性的尝试通过提供更精确的带隙预测和特别与光学特性相关的有价值的见解来实现高性能光电检测系统。
































 京公网安备 11010802027423号
京公网安备 11010802027423号