Science China Materials ( IF 6.8 ) Pub Date : 2023-07-06 , DOI: 10.1007/s40843-022-2501-8 Lei Zhou , Ya-Qiong Su , Tong-Liang Hu
|
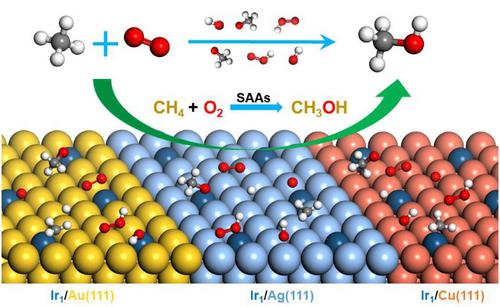
The direct conversion of methane into value-added fuels and chemicals such as methanol has attracted increasing interest, but remains a great challenge due to the chemical inertness of methane. Herein, twelve single-atom alloys (SAAs) were designed for the activation of methane initially, and then the ones owning superior catalytic activity for C-H bond dissociation were screened out for further investigation of selective oxidation of methane to methanol by using density functional theory (DFT) calculations and microkinetic modeling. The results indicate that doping of Ir metal-atom into inert coinage hosts (Ag, Au and Cu) leads to a higher activity for methane dissociation, primarily deriving from the interaction between the C-H bond of methane and the unique projected d-band density of states of the Ir atom. With the introduction of molecular oxygen, three possible reaction mechanisms for selective oxidation of methane to methanol were proposed. Through DFT calculations, the complete reaction networks of three pathways were determined, and microkinetics revealed the changes of surface coverage of reactive species on each SAA under actual reaction conditions. The sophisticated analysis showed oxidation reactions were in favor on Ir1/Ag surface over all temperature ranges while favorable for Ir1/Au and Ir1/Cu within a limited interval, indicating that Ir1/Ag SAA is the most efficient for selective oxidation of methane to methanol. The insights in this work provide important guidelines for the design of highly active and efficient catalysts for direct conversion of methane in future.
中文翻译:
单原子合金催化剂上甲烷选择性氧化为甲醇的理论见解
将甲烷直接转化为增值燃料和甲醇等化学品引起了越来越多的兴趣,但由于甲烷的化学惰性,仍然是一个巨大的挑战。本文首先设计了12种单原子合金(SAA)用于甲烷的活化,然后筛选出对CH键解离具有优异催化活性的单原子合金,以利用密度泛函理论进一步研究甲烷选择性氧化为甲醇的过程( DFT)计算和微动力学建模。结果表明,将 Ir 金属原子掺杂到惰性造币主体(Ag、Au 和 Cu)中会导致更高的甲烷解离活性,这主要源于甲烷的 CH 键与独特的预计 d 带密度之间的相互作用。 Ir 原子的状态。随着分子氧的引入,提出了甲烷选择性氧化成甲醇的三种可能的反应机制。通过DFT计算,确定了三个途径的完整反应网络,微动力学揭示了实际反应条件下各SAA上活性物种表面覆盖度的变化。复杂的分析表明氧化反应有利于 Ir1 /Ag表面在所有温度范围内,而在有限的区间内有利于Ir 1 /Au和Ir 1 /Cu,表明Ir 1 /Ag SAA对于甲烷选择性氧化为甲醇是最有效的。这项工作的见解为未来设计用于甲烷直接转化的高活性和高效催化剂提供了重要指导。






























 京公网安备 11010802027423号
京公网安备 11010802027423号