当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Type II Germanium Clathrates from Zintl Phase Precursor Na4Ge4: Understanding Desodiation Processes and Sodium Migration Using First-Principles Calculations
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-07-05 , DOI: 10.1021/acs.jpcc.3c02343 Anirudh Nandakumar 1 , Xihong Peng 2 , Candace K. Chan 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-07-05 , DOI: 10.1021/acs.jpcc.3c02343 Anirudh Nandakumar 1 , Xihong Peng 2 , Candace K. Chan 1
Affiliation
|
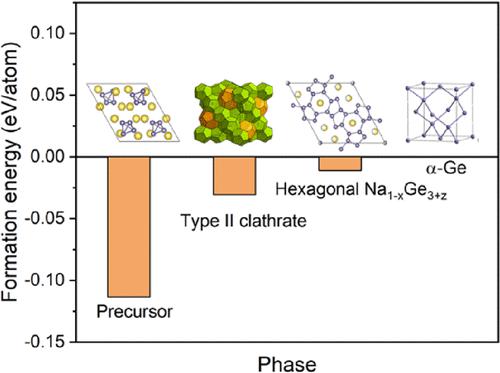
Type II germanium clathrates have recently been investigated for potential applications as anodes in batteries due to their cage-like structures that can accommodate electrochemical insertion of guest ions. To synthesize type II Ge clathrates (Ge136), several experimental routes use thermal or electrochemical desodiation of the Zintl phase compound Na4Ge4. However, the mechanism by which Na atoms are removed from the precursor to form clathrates is not well understood. Herein, we use first-principles density functional theory and nudged elastic band calculations to understand the reaction mechanism and formation energies of the products typically observed in the synthesis, namely, NaδGe136 (0 < δ < 24) type II clathrates and hexagonal phase Na1–xGe3+z. Specifically, we confirm the energetic feasibility of Na vacancy formation in Na4Ge4 and find that the barrier for Na vacancy migration is only 0.37 eV. This relatively low energy barrier is consistent with the ease with which Na4Ge4 can be desodiated to form the products. We also discuss the energetics, sodium migration pathways, and potential electrochemical performance of Ge136 as anode material for Na-ion batteries. Overall, this study highlights how first-principles calculations can be used to understand the synthesis mechanism and desodiation processes in clathrate materials and will help guide researchers in the design and evaluation of new open framework compounds as viable materials for energy storage applications.
中文翻译:

Zintl 相前体 Na4Ge4 的 II 型锗包合物:使用第一原理计算了解脱钠过程和钠迁移
最近,人们研究了 II 型锗包合物作为电池阳极的潜在应用,因为它们的笼状结构可以容纳客体离子的电化学插入。为了合成II型Ge包合物(Ge 136 ),一些实验路线使用Zintl相化合物Na 4 Ge 4的热或电化学脱钠。然而,Na 原子从前体中去除形成包合物的机制尚不清楚。在这里,我们使用第一性原理密度泛函理论和微动弹性带计算来了解合成中通常观察到的产物(即 Na δ Ge 136)的反应机理和形成能。(0 < δ < 24) II 型包合物和六方相 Na 1– x Ge 3+ z。具体来说,我们证实了Na 4 Ge 4中Na空位形成的能量可行性,并发现Na空位迁移的势垒仅为0.37 eV。这种相对较低的能垒与Na 4 Ge 4可以脱钠以形成产物的容易性一致。我们还讨论了 Ge 136的能量学、钠迁移途径和潜在的电化学性能作为钠离子电池的负极材料。总的来说,这项研究强调了如何使用第一性原理计算来理解笼形材料的合成机制和脱钠过程,并将有助于指导研究人员设计和评估新型开放式框架化合物作为储能应用的可行材料。
更新日期:2023-07-05
中文翻译:
Zintl 相前体 Na4Ge4 的 II 型锗包合物:使用第一原理计算了解脱钠过程和钠迁移
最近,人们研究了 II 型锗包合物作为电池阳极的潜在应用,因为它们的笼状结构可以容纳客体离子的电化学插入。为了合成II型Ge包合物(Ge 136 ),一些实验路线使用Zintl相化合物Na 4 Ge 4的热或电化学脱钠。然而,Na 原子从前体中去除形成包合物的机制尚不清楚。在这里,我们使用第一性原理密度泛函理论和微动弹性带计算来了解合成中通常观察到的产物(即 Na δ Ge 136)的反应机理和形成能。(0 < δ < 24) II 型包合物和六方相 Na 1– x Ge 3+ z。具体来说,我们证实了Na 4 Ge 4中Na空位形成的能量可行性,并发现Na空位迁移的势垒仅为0.37 eV。这种相对较低的能垒与Na 4 Ge 4可以脱钠以形成产物的容易性一致。我们还讨论了 Ge 136的能量学、钠迁移途径和潜在的电化学性能作为钠离子电池的负极材料。总的来说,这项研究强调了如何使用第一性原理计算来理解笼形材料的合成机制和脱钠过程,并将有助于指导研究人员设计和评估新型开放式框架化合物作为储能应用的可行材料。
































 京公网安备 11010802027423号
京公网安备 11010802027423号