Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2023-07-02 , DOI: 10.1002/adsc.202300252
Juan Diego Pizarro 1 , Lucia Moran-Gonzalez 2 , Ivan Gonzalez-Fernandez 3 , Feliu Maseras 4 , Manuel R. Fructos 5 , Pedro J. Perez 3
|
Introduction
The transfer of carbene units from diazo compounds catalyzed by transition metal complexes constitutes a valuable tool in organic synthesis.1 Among the plethora of reactions described within this methodology, the functionalization of C−H bonds upon insertion of that carbene unit is one of the most interesting targets.2 Its application to indole substrates has gained interest in the last decade,3 due to the presence of this structure in natural compounds and to their uses in the pharmaceutical and agrochemical industries.4 Scheme 1 shows the different examples known to date for the modification of indoles by carbene transfer reactions: (a) cyclopropanation of the C=C bond;5 (b) N−H functionalization;6 (c) C2−H functionalization7 and (d) C3−H functionalization.8 Occasionally, the six-member ring can also be modified.5e The presence of different reaction sites frequently generate selectivity issues. The preferred derivatives are those from the C2−H and C3−H sites. Most of the work toward that end has been carried out employing N-protected indoles, therefore eliminating the drawback of the N−H functionalization, which is by far the most reactive site. Systems selective for C−H bond functionalization by carbene insertion tolerant with the N−H functionality are scarce: Koenigs9 has described the incorporation of carbene units to C−H bonds of unprotected indoles and carbazoles, whereas Fasan and Arnold have employed more elaborated catalysts based on myoglobin or cytochrome P450, respectively, toward that end.10 Therefore the direct C−H bond functionalization of N−H indoles with this strategy yet constitutes a challenge in this area.

Functionalization of indoles by transition metal-catalyzed carbene transfer from diazocompounds.
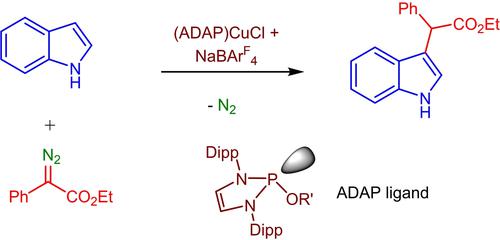
Previous work in our laboratory showed that the complexes TpxCu(NCMe) (Tpx=hydrotrispyrazolylborate ligand) display good catalytic properties for the functionalization of the C3−H position of protected indoles by carbene insertion from acceptor diazo compounds. However, with unprotected indoles the chemoselectivity was low, generating mixtures of products from C3−H and N−H bond functionalization.[8b] Based on the scarcity of catalytic systems for the modification of C−H bonds of unprotected indoles, we have now targeted such goal. Herein we describe a novel family of copper catalysts containing alkoxydiaminophosphine (ADAP) ligands for the selective functionalization of unprotected indoles at the C3−H position using donor-acceptor diazo compounds, leaving the N−H group unreacted.
中文翻译:
通过供体-受体卡宾插入选择性 C−H 键功能化未受保护的吲哚
介绍
由过渡金属配合物催化的重氮化合物的卡宾单元的转移构成了有机合成中的有价值的工具。1在该方法描述的众多反应中,插入卡宾单元后 C−H 键的官能化是最有趣的目标之一。2过去十年,它在吲哚底物中的应用引起了人们的兴趣,3由于天然化合物中存在这种结构及其在制药和农用化学工业中的用途。4方案 1 显示了迄今为止已知的通过卡宾转移反应修饰吲哚的不同实例: (a) C=C 键的环丙烷化;5 (b) N−H 官能化;6 (c) C2-H 官能化7和 (d) C3-H 官能化。8有时,六元环也可以进行修改。5e不同反应位点的存在经常会产生选择性问题。优选的衍生物是来自C2-H和C3-H位点的那些。为此目的的大部分工作都是使用 N-保护的吲哚进行的,因此消除了 NH 官能化的缺点,这是迄今为止最具反应性的位点。通过卡宾插入选择性地进行 C−H 键官能化的系统很少,并且能够容忍 N−H 官能团:Koenigs 9描述了卡宾单元与未受保护的吲哚和咔唑的 C−H 键的结合,而 Fasan 和 Arnold 采用了更复杂的催化剂为此,分别基于肌红蛋白或细胞色素 P450。10因此,采用这种策略对 N−H 吲哚进行直接 C−H 键功能化仍构成该领域的挑战。
通过过渡金属催化的重氮化合物的卡宾转移对吲哚进行官能化。
我们实验室之前的工作表明,配合物Tp x Cu(NCMe)(Tp x =氢化三吡唑基硼酸配体)通过受体重氮化合物的卡宾插入对受保护吲哚的C3−H位置进行官能化显示出良好的催化性能。然而,对于未受保护的吲哚,化学选择性较低,会产生 C3−H 和 N−H 键功能化产物的混合物。[8b]基于未受保护的吲哚 C−H 键修饰的催化系统的稀缺性,我们现在有了瞄准了这样的目标。在此,我们描述了一种含有烷氧基二氨基膦 (ADAP) 配体的新型铜催化剂,用于使用供体-受体重氮化合物在 C3−H 位上选择性官能化未受保护的吲哚,而使 NH 基团不反应。
































 京公网安备 11010802027423号
京公网安备 11010802027423号