European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2023-07-01 , DOI: 10.1016/j.ejmech.2023.115621 Guoqing Fang 1 , Hongjuan Chen 1 , Zhiyun Cheng 1 , Zilong Tang 1 , Yichao Wan 1
|
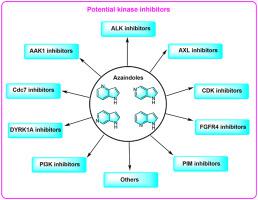
Currently, heterocycles have occupied an important position in the fields of drug design. Among them, azaindole moiety is regarded as one privileged scaffold to develop therapeutic agents. Since two nitrogen atoms of azaindole increase the possibility to form hydrogen bonds in the adenosine triphosphate (ATP)-binding site, azaindole derivatives are important sources of kinase inhibitors. Moreover, some of them have been on the market or in clinical trials for the treatment of some kinase-related diseases (e.g., vemurafenib, pexidartinib, decernotinib). In this review, we focused on the recent development of azaindole derivatives as potential kinase inhibitors based on kinase targets, such as adaptor-associated kinase 1 (AAK1), anaplastic lymphoma kinase (ALK), AXL, cell division cycle 7 (Cdc7), cyclin-dependent kinases (CDKs), dual-specificity tyrosine (Y)-phosphorylation regulated kinase 1A (DYRK1A), fibroblast growth factor receptor 4 (FGFR4), phosphatidylinositol 3-kinase (PI3K) and proviral insertion site in moloney murine leukemia virus (PIM) kinases. Meanwhile, the structure-activity relationships (SARs) of most azaindole derivatives were also elucidated. In addition, the binding modes of some azaindoles complexed with kinases were also investigated during the SARs elucidation. This review may offer an insight for medicinal chemists to rationally design more potent kinase inhibitors bearing the azaindole scaffold.
中文翻译:
作为潜在激酶抑制剂的氮杂吲哚衍生物及其 SAR 阐明
目前,杂环化合物在药物设计领域占据了重要地位。其中,氮杂吲哚部分被认为是开发治疗药物的一种优先支架。由于氮杂吲哚的两个氮原子增加了在三磷酸腺苷(ATP)结合位点形成氢键的可能性,因此氮杂吲哚衍生物是激酶抑制剂的重要来源。而且,其中一些已经上市或处于临床试验阶段,用于治疗一些激酶相关疾病(例如vemurafenib、 pexidartinib 、decernotinib)。在这篇综述中,我们重点关注了氮杂吲哚衍生物作为基于激酶靶点的潜在激酶抑制剂的最新发展,例如接头相关激酶 1 (AAK1)、间变性淋巴瘤激酶 (ALK)、AXL、细胞分裂周期 7 (Cdc7) 、细胞周期蛋白依赖性激酶 (CDK)、双特异性酪氨酸(Y) 磷酸化调节激酶 1A (DYRK1A)、成纤维细胞生长因子受体 4 (FGFR4)、磷脂酰肌醇 3-激酶 (PI3K) 和莫洛尼鼠白血病病毒中的前病毒插入位点( PIM) 激酶。同时,大多数氮杂吲哚衍生物的构效关系(SAR)也得到了阐明。此外,在SAR阐明过程中还研究了一些与激酶复合的氮杂吲哚的结合模式。该综述可能为药物化学家合理设计带有氮杂吲哚支架的更有效的激酶抑制剂提供见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号