Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Discovery of deaminase functions by structure-based protein clustering
Cell ( IF 45.5 ) Pub Date : 2023-06-27 , DOI: 10.1016/j.cell.2023.05.041 Jiaying Huang 1 , Qiupeng Lin 1 , Hongyuan Fei 2 , Zixin He 2 , Hu Xu 3 , Yunjia Li 2 , Kunli Qu 4 , Peng Han 4 , Qiang Gao 3 , Boshu Li 2 , Guanwen Liu 1 , Lixiao Zhang 3 , Jiacheng Hu 1 , Rui Zhang 1 , Erwei Zuo 5 , Yonglun Luo 6 , Yidong Ran 3 , Jin-Long Qiu 7 , Kevin Tianmeng Zhao 3 , Caixia Gao 2
Cell ( IF 45.5 ) Pub Date : 2023-06-27 , DOI: 10.1016/j.cell.2023.05.041 Jiaying Huang 1 , Qiupeng Lin 1 , Hongyuan Fei 2 , Zixin He 2 , Hu Xu 3 , Yunjia Li 2 , Kunli Qu 4 , Peng Han 4 , Qiang Gao 3 , Boshu Li 2 , Guanwen Liu 1 , Lixiao Zhang 3 , Jiacheng Hu 1 , Rui Zhang 1 , Erwei Zuo 5 , Yonglun Luo 6 , Yidong Ran 3 , Jin-Long Qiu 7 , Kevin Tianmeng Zhao 3 , Caixia Gao 2
Affiliation
|
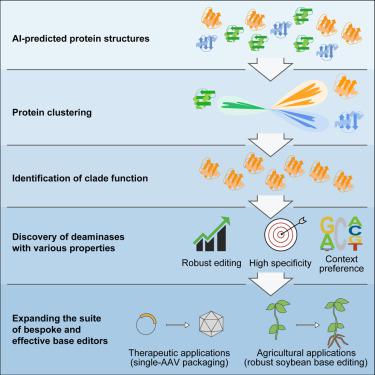
The elucidation of protein function and its exploitation in bioengineering have greatly advanced the life sciences. Protein mining efforts generally rely on amino acid sequences rather than protein structures. We describe here the use of AlphaFold2 to predict and subsequently cluster an entire protein family based on predicted structure similarities. We selected deaminase proteins to analyze and identified many previously unknown properties. We were surprised to find that most proteins in the DddA-like clade were not double-stranded DNA deaminases. We engineered the smallest single-strand-specific cytidine deaminase, enabling efficient cytosine base editor (CBE) to be packaged into a single adeno-associated virus (AAV). Importantly, we profiled a deaminase from this clade that edits robustly in soybean plants, which previously was inaccessible to CBEs. These discovered deaminases, based on AI-assisted structural predictions, greatly expand the utility of base editors for therapeutic and agricultural applications.
中文翻译:

通过基于结构的蛋白质聚类发现脱氨酶功能
蛋白质功能的阐明及其在生物工程中的开发极大地推进了生命科学。蛋白质挖掘工作通常依赖于氨基酸序列而不是蛋白质结构。我们在这里描述了使用 AlphaFold2 来预测并随后根据预测的结构相似性对整个蛋白质家族进行聚类。我们选择脱氨酶蛋白来分析和鉴定许多以前未知的特性。我们惊讶地发现 DddA 样进化枝中的大多数蛋白质都不是双链 DNA 脱氨酶。我们设计了最小的单链特异性胞苷脱氨酶,使有效的胞嘧啶碱基编辑器(CBE)能够包装到单个腺相关病毒(AAV)中。重要的是,我们分析了该进化枝中的一种脱氨酶,它可以在大豆植物中进行强大的编辑,而这种酶以前是 CBE 无法接触到的。这些基于人工智能辅助结构预测的脱氨酶极大地扩展了碱基编辑器在治疗和农业应用中的实用性。
更新日期:2023-06-27
中文翻译:
通过基于结构的蛋白质聚类发现脱氨酶功能
蛋白质功能的阐明及其在生物工程中的开发极大地推进了生命科学。蛋白质挖掘工作通常依赖于氨基酸序列而不是蛋白质结构。我们在这里描述了使用 AlphaFold2 来预测并随后根据预测的结构相似性对整个蛋白质家族进行聚类。我们选择脱氨酶蛋白来分析和鉴定许多以前未知的特性。我们惊讶地发现 DddA 样进化枝中的大多数蛋白质都不是双链 DNA 脱氨酶。我们设计了最小的单链特异性胞苷脱氨酶,使有效的胞嘧啶碱基编辑器(CBE)能够包装到单个腺相关病毒(AAV)中。重要的是,我们分析了该进化枝中的一种脱氨酶,它可以在大豆植物中进行强大的编辑,而这种酶以前是 CBE 无法接触到的。这些基于人工智能辅助结构预测的脱氨酶极大地扩展了碱基编辑器在治疗和农业应用中的实用性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号