当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural Evolution of Paramagnetic Lanthanide Compounds in Solution Compared to Time- and Ensemble-Average Structures
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-06-16 , DOI: 10.1021/jacs.3c01342 Barak Alnami 1 , Jon G C Kragskow 1 , Jakob K Staab 1 , Jonathan M Skelton 1 , Nicholas F Chilton 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-06-16 , DOI: 10.1021/jacs.3c01342 Barak Alnami 1 , Jon G C Kragskow 1 , Jakob K Staab 1 , Jonathan M Skelton 1 , Nicholas F Chilton 1
Affiliation
|
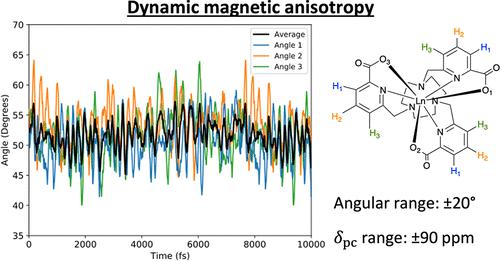
Anisotropy in the magnetic susceptibility strongly influences the paramagnetic shifts seen in nuclear magnetic resonance (NMR) and magnetic resonance imaging (MRI) experiments. A previous study on a series of C3-symmetric prototype MRI contrast agents showed that their magnetic anisotropy was highly sensitive to changes in molecular geometry and concluded that changes in the average angle between the lanthanide–oxygen (Ln–O) bonds and the molecular C3 axis due to solvent interactions had a significant impact on the magnetic anisotropy and, consequently, the paramagnetic shift. However, this study, like many others, was predicated on an idealized C3-symmetric structural model, which may not be representative of the dynamic structure in solution at the single-molecule level. Here, we address this by using ab initio molecular dynamics simulations to simulate how the molecular geometry, in particular the angles between the Ln–O bonds and the pseudo-C3 axis, evolves over time in the solution, mimicking typical experimental conditions. We observe large-amplitude oscillations in the O–Ln–C̃3 angles, and complete active space self-consistent field spin–orbit calculations show that this leads to similarly large oscillations in the pseudocontact (dipolar) paramagnetic NMR shifts. The time-averaged shifts show good agreement with experimental measurements, while the large fluctuations suggest that an idealized structure provides an incomplete description of the solution dynamics. Our observations have significant implications for modeling the electronic and nuclear relaxation times in this and other systems where the magnetic susceptibility is exquisitely sensitive to the molecular structure.
中文翻译:

溶液中顺磁稀土化合物的结构演化与时间平均和系综平均结构的比较
磁化率的各向异性强烈影响核磁共振 (NMR) 和磁共振成像 (MRI) 实验中观察到的顺磁位移。先前对一系列 C 3对称原型 MRI 造影剂的研究表明,它们的磁各向异性对分子几何形状的变化高度敏感,并得出结论,镧系元素 - 氧 (Ln-O) 键和分子之间的平均角度的变化由于溶剂相互作用而产生的C 3轴对磁各向异性以及顺磁位移具有显着影响。然而,与许多其他研究一样,这项研究是以理想化的 C 3对称结构模型为基础的,该模型可能无法代表单分子水平上溶液中的动态结构。在这里,我们通过使用从头算分子动力学模拟来模拟分子几何形状(特别是 Ln-O 键和伪 C 3轴之间的角度)如何在溶液中随时间演变,从而模拟典型的实验条件来解决这个问题。我们观察到 O-Ln-C̃ 3角的大幅振荡,并且完整的活动空间自洽场自旋轨道计算表明,这导致赝接触(偶极)顺磁 NMR 位移中出现类似的大振荡。时间平均位移与实验测量结果吻合良好,而较大的波动表明理想化结构无法完整地描述溶液动力学。我们的观察结果对于模拟该系统和其他系统中的电子和核弛豫时间具有重要意义,其中磁化率对分子结构非常敏感。
更新日期:2023-06-16
中文翻译:
溶液中顺磁稀土化合物的结构演化与时间平均和系综平均结构的比较
磁化率的各向异性强烈影响核磁共振 (NMR) 和磁共振成像 (MRI) 实验中观察到的顺磁位移。先前对一系列 C 3对称原型 MRI 造影剂的研究表明,它们的磁各向异性对分子几何形状的变化高度敏感,并得出结论,镧系元素 - 氧 (Ln-O) 键和分子之间的平均角度的变化由于溶剂相互作用而产生的C 3轴对磁各向异性以及顺磁位移具有显着影响。然而,与许多其他研究一样,这项研究是以理想化的 C 3对称结构模型为基础的,该模型可能无法代表单分子水平上溶液中的动态结构。在这里,我们通过使用从头算分子动力学模拟来模拟分子几何形状(特别是 Ln-O 键和伪 C 3轴之间的角度)如何在溶液中随时间演变,从而模拟典型的实验条件来解决这个问题。我们观察到 O-Ln-C̃ 3角的大幅振荡,并且完整的活动空间自洽场自旋轨道计算表明,这导致赝接触(偶极)顺磁 NMR 位移中出现类似的大振荡。时间平均位移与实验测量结果吻合良好,而较大的波动表明理想化结构无法完整地描述溶液动力学。我们的观察结果对于模拟该系统和其他系统中的电子和核弛豫时间具有重要意义,其中磁化率对分子结构非常敏感。












































 京公网安备 11010802027423号
京公网安备 11010802027423号