当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
金属有机框架中 CO2 吸附热力学函数的从头计算:横向相互作用的熵效应
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-06-12 , DOI: 10.1021/acs.jpcc.3c02234 Kaido Sillar 1 , Ivar Koppel 2
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-06-12 , DOI: 10.1021/acs.jpcc.3c02234 Kaido Sillar 1 , Ivar Koppel 2
Affiliation
|
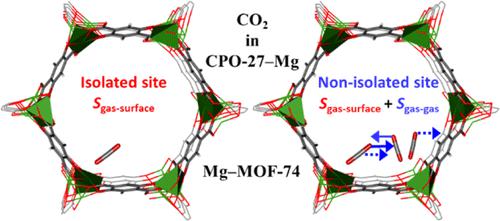
我们使用量子化学与分子统计学相结合来计算二氧化碳与金属有机骨架 Mg 2 (dobdc)(也称为CPO -27-Mg 和 Mg)中开放金属位点结合的吉布斯自由能、焓和吸附熵。 –MOF-74。对于气体-表面相互作用能,我们将周期 MP2 和 DFT+D 结合起来形成全晶体框架,并结合 CCSD(T) 形成吸附位点。热力学函数是根据隔离位点的非谐振动频率以及吸附 CO 2的较高表面覆盖度计算的。分子与邻近的分子相互作用。分子间横向振动带来了一组额外的构型,这些构型在分子处于孤立位置时不存在。横向吸附质-吸附质相互作用的贡献可以单独计算,并将它们考虑在内,结果是实验等温线和从头计算得出的热力学函数之间具有良好的一致性(±1 kJ/mol)。

"点击查看英文标题和摘要"
更新日期:2023-06-12
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号