Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy ( IF 4.3 ) Pub Date : 2023-06-08 , DOI: 10.1016/j.saa.2023.122988 Ranjith Balu 1 , Anthoniammal Panneerselvam 2 , Gautham Devendrapandi 3 , Jothi Ramalingam Rajabathar 4 , Hamad A Al-Lohedan 4 , Dhaifallah M Al-Dhayan 4
|
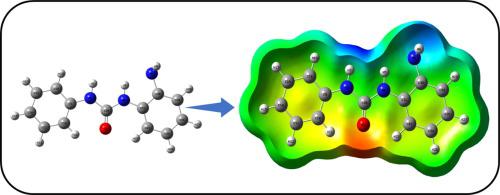
The present study focuses on structural and chemical analyses of N-phenylmorpholine-4-carboxamide benzene-1,2-diamine (PMCBD) using quantum computational methods. The calculated bond angle, length, and dihedral angle between atoms were compared with measured values. The observed and stimulated FT-IR (Fourier Transform Infrared Spectroscopy) spectra parameters for vibrational wavenumbers and their associated PED (Potential Energy Distribution) values in percentage have been obtained from VEDA4 software. The electronic transitions of PMCBD were discussed by TD-SCF/DFT/B3LYP based on the 6-311++G(d,p) basis set with solvents such as chloroform, ethanol, and dimethyl sulfoxide (DMSO) and gas. Density functional computations were used to study the band energy between HOMO and LUMO using the B3LYP/6-311++G(d,p) level. Mulliken analysis and natural population analysis were used for a better understanding of charge levels on different atoms such as N, H and O. The natural bonding orbital (NBO) analysis proved helpful in studying molecular and bond strengths. (NBO). The ESP acquired data on the molecule's size, shape, charge density distribution, and chemical reactivity site. This was done by mapping electron density on the surface with electrostatic potential. Non-linear optical detection of PMCBD was also discussed. Aside from the electron localization function map, state densities are also mapped using Multiwfn software, a wave function analyzer.
中文翻译:
N-苯基吗啉-4-甲酰胺苯-1,2-二胺波函数的理论和实验光谱研究与分析与计算技术
本研究的重点是使用量子计算方法对 N-苯基吗啉-4-甲酰胺苯-1,2-二胺 (PMCBD) 进行结构和化学分析。将计算的键角、长度和原子间的二面角与测量值进行比较。已从 VEDA4 软件获得振动波数的观察和受激 FT-IR(傅立叶变换红外光谱)光谱参数及其相关的 PED(势能分布)值(以百分比表示)。PMCBD 的电子跃迁通过 TD-SCF/DFT/B3LYP 基于 6-311++G(d,p) 基组与溶剂如氯仿、乙醇和二甲基亚砜 (DMSO) 和气体进行了讨论。密度泛函计算用于使用 B3LYP/6-311++G(d,p) 能级研究 HOMO 和 LUMO 之间的能带。Mulliken 分析和自然布居分析用于更好地了解不同原子(例如 N、H 和 O)的电荷水平。事实证明,自然键合轨道 (NBO) 分析有助于研究分子和键强度。(NBO)。ESP 获取了有关分子大小、形状、电荷密度分布和化学反应位点的数据。这是通过用静电势映射表面上的电子密度来完成的。还讨论了 PMCBD 的非线性光学检测。除了电子局域化函数图外,状态密度也使用 Multiwfn 软件(一种波函数分析仪)进行映射。ESP 获取了有关分子大小、形状、电荷密度分布和化学反应位点的数据。这是通过用静电势映射表面上的电子密度来完成的。还讨论了 PMCBD 的非线性光学检测。除了电子局域化函数图外,状态密度也使用 Multiwfn 软件(一种波函数分析仪)进行映射。ESP 获取了有关分子大小、形状、电荷密度分布和化学反应位点的数据。这是通过用静电势映射表面上的电子密度来完成的。还讨论了 PMCBD 的非线性光学检测。除了电子局域化函数图外,状态密度也使用 Multiwfn 软件(一种波函数分析仪)进行映射。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号