Applied Surface Science ( IF 6.3 ) Pub Date : 2023-05-18 , DOI: 10.1016/j.apsusc.2023.157534 Yuchen Wang , Liang Zhu , Yang Liu , Evgeny I. Vovk , Junyu Lang , Zixuan Zhou , Peng Gao , Shenggang Li , Yong Yang
|
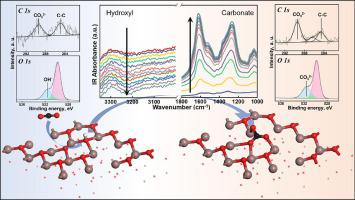
The In2O3 catalyst has been shown to have a high activity for CO2 hydrogenation to methanol. For this high pressure process, reliable spectral assignment of surface species formed by interaction with CO2, H2O, and other major reaction ingredients, is the key step to achieve mechanistic understanding. In this study, in situ IR and XPS are performed to investigate the O1s binding energies of the adsorbates induced by CO2 and H2O treatments along with density functional theory (DFT) simulations to further correlate the calibrated assignments with surface structures. Time resolved IR indicates that carbonates formation induced by CO2 exposure replaces the original hydroxyl on In2O3, and XPS further reveals that these two oxygen species have very similar binding energies (532.0 ± 0.2 eV). Computational results using the In2O3(1 1 0) and (1 1 1) slab models give good agreement with the XPS experiments, further suggesting that the amount of oxygen vacancy concentration does not induce new lattice oxygen O1s binding energy, but resulting in a slight shift to a higher binding energy instead. Our results provide useful structure information for the In2O3 catalyst surface and shed light for further investigations into its kinetic behavior under methanol synthesis condition.
中文翻译:
使用时间分辨红外、原位处理的 XPS 和 DFT 计算了解 CO2 加氢反应过程中 In2O3 催化剂的表面结构
In 2 O 3催化剂已被证明对CO 2氢化成甲醇具有高活性。对于这种高压过程,通过与 CO 2、H 2 O 和其他主要反应成分相互作用形成的表面物质的可靠光谱分配是实现机理理解的关键步骤。在这项研究中,执行原位IR 和 XPS 以研究由 CO 2和 H 2 O 处理诱导的吸附物的 O1s 结合能以及密度泛函理论 (DFT) 模拟,以进一步将校准分配与表面结构相关联。时间分辨 IR 表明 CO 诱导碳酸盐形成2暴露取代了 In 2 O 3上的原始羟基,XPS 进一步揭示这两种氧具有非常相似的结合能 (532.0 ± 0.2 eV)。使用 In 2 O 3 (1 1 0) 和 (1 1 1) 平板模型的计算结果与 XPS 实验非常吻合,进一步表明氧空位浓度的数量不会引起新的晶格氧 O1s 结合能,但会导致相反,它略微转向更高的结合能。我们的结果为 In 2 O 3提供了有用的结构信息催化剂表面并阐明进一步研究其在甲醇合成条件下的动力学行为。
































 京公网安备 11010802027423号
京公网安备 11010802027423号