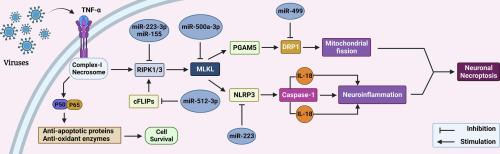
中枢神经系统中的神经元坏死(程序性坏死)通过不依赖半胱天冬酶的方式自然发生,特别是在神经退行性疾病(NDD)中,例如阿尔茨海默病(AD)、帕金森病(PD)、肌萎缩侧索硬化症(ALS)和病毒感染。了解坏死性凋亡途径(死亡受体依赖性和独立性)及其与其他细胞死亡途径的联系可能会给治疗带来新的见解。受体相互作用蛋白激酶 (RIPK) 通过混合谱系激酶样 (MLKL) 蛋白介导坏死性凋亡。RIPK/MLKL 坏死体含有 FADD、procaspase-8 细胞 FLICE 抑制蛋白 (cFLIP)、RIPK1/RIPK3 和 MLKL。坏死刺激导致 MLKL 磷酸化并易位至质膜,导致 Ca 2+和 Na +离子流入,线粒体通透性转换孔 (mPTP) 立即打开,释放炎症细胞损伤相关分子模式。 DAMP)如线粒体 DNA (mtDNA)、高迁移率族蛋白 1 (HMGB1) 和白细胞介素 1 (IL-1)。MLKL 易位至细胞核,诱导 NLRP3 炎性体复合体元件的转录。MLKL 诱导的 NLRP3 活性导致 caspase-1 裂解和 IL-1 激活,从而促进神经炎症。RIPK1 依赖性转录会增加与疾病相关的小胶质细胞和溶酶体异常,从而促进 AD 中淀粉样斑块 (Aβ) 的聚集。最近的研究将神经炎症和线粒体裂变与坏死性凋亡联系起来。miR512-3p、miR874、miR499、miR155 和 miR128a 等 MicroRNA (miR) 通过靶向坏死性凋亡途径的关键成分来调节神经元坏死性凋亡。坏死性凋亡抑制剂通过抑制 MLKL 和 RIPK1 活性的膜易位发挥作用。本综述深入探讨了死亡受体依赖性和独立性神经元坏死性凋亡过程中 RIPK/MLKL 坏死体-NLRP3 炎性体的相互作用,以及通过 miR 进行临床干预以保护大脑免受 NDD 的影响。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Viral-induced neuronal necroptosis: Detrimental to brain function and regulation by necroptosis inhibitors
Neuronal necroptosis (programmed necrosis) in the CNS naturally occurs through a caspase-independent way and, especially in neurodegenerative diseases (NDDs) such as Alzheimer’s disease (AD), Parknson’s disease (PD), Amyotrophic Lateral Sclerosis (ALS) and viral infections. Understanding necroptosis pathways (death receptor-dependent and independent), and its connections with other cell death pathways could lead to new insights into treatment. Receptor-interacting protein kinase (RIPK) mediates necroptosis via mixed-lineage kinase-like (MLKL) proteins. RIPK/MLKL necrosome contains FADD, procaspase-8-cellular FLICE-inhibitory proteins (cFLIPs), RIPK1/RIPK3, and MLKL. The necrotic stimuli cause phosphorylation of MLKL and translocate to the plasma membrane, causing an influx of Ca2+ and Na+ ions and, the immediate opening of mitochondrial permeability transition pore (mPTP) with the release of inflammatory cell damage-associated molecular patterns (DAMPs) like mitochondrial DNA (mtDNA), high-mobility group box1 (HMGB1), and interleukin1 (IL-1). The MLKL translocates to the nucleus to induce transcription of the NLRP3 inflammasome complex elements. MLKL-induced NLRP3 activity causes caspase-1 cleavage and, IL-1 activation which promotes neuroinflammation. RIPK1-dependent transcription increases illness-associated microglial and lysosomal abnormalities to facilitate amyloid plaque (Aβ) aggregation in AD. Recent research has linked neuroinflammation and mitochondrial fission with necroptosis. MicroRNAs (miRs) such as miR512-3p, miR874, miR499, miR155, and miR128a regulate neuronal necroptosis by targeting key components of necroptotic pathways. Necroptosis inhibitors act by inhibiting the membrane translocation of MLKL and RIPK1 activity. This review insights into the RIPK/MLKL necrosome-NLRP3 inflammasome interactions during death receptor-dependent and independent neuronal necroptosis, and clinical intervention by miRs to protect the brain from NDDs.




















































 京公网安备 11010802027423号
京公网安备 11010802027423号