Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2023-04-13 , DOI: 10.1016/j.molstruc.2023.135572 K. Srishailam , A. Balakrishna , B. Venkatram Reddy , G. Ramana Rao
|
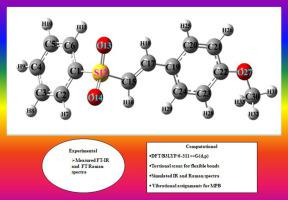
Fourier transform infrared and Raman spectra, in the spectral range 4000- 400 and 4000–50 cm−1, respectively, were measured for 1‑methoxy-4-[2-(phenylsulfonyl)vinyl]benzene (MPB). Initial values of torsion angles around five flexible bonds C1-S, S-C15, C17-C19, C22-O and O C30 (see Fig. 1 for numbering), essential for starting geometry optimization, were determined using "bond-pairing method" proposed earlier by us. Optimized structure parameters, harmonic general valence force field, vibrational fundamentals, potential energy distribution and infrared and Raman band intensities were determined using density functional theory, employing B3LYP/ 6–311++G(d,p) formalism. Good agreement was found, between measured and computed quantities investigated here. The r.m.s error between experimental and theoretical vibrational wavenumbers was 8.6 cm−1, for MPB. With the help of PED and eigenvectors, all vibrational fundamentals of the molecule were assigned unambiguously for the first time.
C30 (see Fig. 1 for numbering), essential for starting geometry optimization, were determined using "bond-pairing method" proposed earlier by us. Optimized structure parameters, harmonic general valence force field, vibrational fundamentals, potential energy distribution and infrared and Raman band intensities were determined using density functional theory, employing B3LYP/ 6–311++G(d,p) formalism. Good agreement was found, between measured and computed quantities investigated here. The r.m.s error between experimental and theoretical vibrational wavenumbers was 8.6 cm−1, for MPB. With the help of PED and eigenvectors, all vibrational fundamentals of the molecule were assigned unambiguously for the first time.
中文翻译:
深入了解 1-methoxy-4-[2-(phenylsulfonyl)vinyl]benzene 的结构和振动特征:实验振动光谱和密度泛函理论的应用
傅立叶变换红外和拉曼光谱分别在 4000-400 和 4000-50 cm -1的光谱范围内测量 1-methoxy-4-[2-( phenylsulfonyl ) vinyl ]benzene (MPB)。围绕五个柔性键 C1-S、S-C15、C17-C19、C22-O 和 O C30(编号见图 1)的扭转角初始值,对于开始几何优化至关重要,使用“键配对方法”确定“我们早先提出的。使用密度泛函理论,采用 B3LYP/ 6–311++ G确定优化的结构参数、谐波一般价力场、振动基本原理、势能分布以及红外和拉曼带强度(d,p) 形式主义。在此处调查的测量量和计算量之间发现了良好的一致性。对于 MPB ,实验和理论振动波数之间的均方根误差为 8.6 cm -1。在 PED 和特征向量的帮助下,分子的所有振动基本原理首次被明确分配。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号