当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DFT Study of the NO Reduction Mechanism on Ag/γ-Al2O3 Catalysts
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-04-11 , DOI: 10.1021/acs.jpcc.2c09042
Ekaterina G. Ragoyja 1 , Vitaly E. Matulis 1 , Oleg A. Ivashkevich 2 , Dmitry A. Lyakhov 3 , Dominik Michels 3
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-04-11 , DOI: 10.1021/acs.jpcc.2c09042
Ekaterina G. Ragoyja 1 , Vitaly E. Matulis 1 , Oleg A. Ivashkevich 2 , Dmitry A. Lyakhov 3 , Dominik Michels 3
Affiliation
|
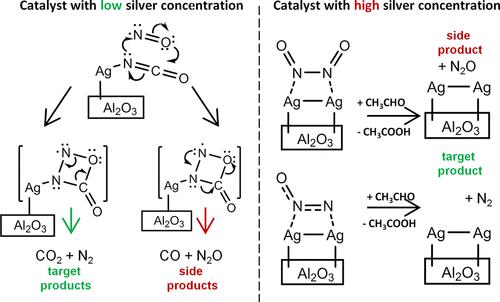
NO catalytic reduction on Ag/γ-Al2O3 catalysts is a very promising process from the industrial and ecological perspective. Details of its mechanism, which are still not fully clear, have great importance for a deep understanding of various heterogeneous NO reduction processes. In this work, a thorough theoretical study of the mechanism of NO reduction on the Ag/γ-Al2O3 catalyst is carried out. Two schemes of the mechanism for catalysts with different silver concentrations and, subsequently, with different reaction centers, are proposed. For the catalyst with a low silver content, a mechanism based on isocyanate species is proposed, while for catalysts with a high silver content, key intermediates are adsorbed NO dimers. The thermodynamic and kinetic feasibility of the proposed schemes is confirmed by density functional theory calculations of the reaction pathways both on isolated silver clusters and on the catalyst surface. These schemes explain the experimentally observed N2O or N2 prevalence in the reaction products. Calculations of the catalyst surface are carried out within the original three-layer embedded cluster model, which provides accurate results of calculations of vibrational frequencies, geometries, and energy characteristics. The process of silver particle migration along the catalyst surface is studied. Energy barriers of migration are estimated. The influence of the catalytic center nature and presence of the aluminum oxide support on NO, N2, and N2O adsorption processes are studied, and the corresponding adsorption energies are calculated.
中文翻译:

Ag/γ-Al2O3 催化剂上 NO 还原机理的 DFT 研究
从工业和生态学的角度来看, Ag/γ-Al 2 O 3催化剂上的 NO 催化还原是一个非常有前途的过程。其机制的细节尚不完全清楚,但对于深入了解各种非均相 NO 还原过程具有重要意义。在这项工作中,对 Ag/γ-Al 2 O 3上 NO 还原机理的深入理论研究催化剂进行。提出了两种具有不同银浓度和随后具有不同反应中心的催化剂的机理方案。对于银含量低的催化剂,提出了基于异氰酸酯物种的机理,而对于银含量高的催化剂,关键中间体是吸附的 NO 二聚体。所提出方案的热力学和动力学可行性通过对分离的银团簇和催化剂表面上的反应路径的密度泛函理论计算得到证实。这些方案解释了实验观察到的 N 2 O 或 N 2普遍存在于反应产物中。催化剂表面的计算是在原始的三层嵌入式簇模型中进行的,它提供了准确的振动频率、几何形状和能量特性的计算结果。研究了银粒子沿催化剂表面迁移的过程。估计迁移的能量障碍。研究了催化中心性质和氧化铝载体的存在对NO、N 2和N 2 O吸附过程的影响,并计算了相应的吸附能。
更新日期:2023-04-11
中文翻译:
Ag/γ-Al2O3 催化剂上 NO 还原机理的 DFT 研究
从工业和生态学的角度来看, Ag/γ-Al 2 O 3催化剂上的 NO 催化还原是一个非常有前途的过程。其机制的细节尚不完全清楚,但对于深入了解各种非均相 NO 还原过程具有重要意义。在这项工作中,对 Ag/γ-Al 2 O 3上 NO 还原机理的深入理论研究催化剂进行。提出了两种具有不同银浓度和随后具有不同反应中心的催化剂的机理方案。对于银含量低的催化剂,提出了基于异氰酸酯物种的机理,而对于银含量高的催化剂,关键中间体是吸附的 NO 二聚体。所提出方案的热力学和动力学可行性通过对分离的银团簇和催化剂表面上的反应路径的密度泛函理论计算得到证实。这些方案解释了实验观察到的 N 2 O 或 N 2普遍存在于反应产物中。催化剂表面的计算是在原始的三层嵌入式簇模型中进行的,它提供了准确的振动频率、几何形状和能量特性的计算结果。研究了银粒子沿催化剂表面迁移的过程。估计迁移的能量障碍。研究了催化中心性质和氧化铝载体的存在对NO、N 2和N 2 O吸附过程的影响,并计算了相应的吸附能。

































 京公网安备 11010802027423号
京公网安备 11010802027423号