当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
使用第一性原理计算优化 SnSe2 的热电性质
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-03-31 , DOI: 10.1021/acs.jpcc.2c09137
Sree Sourav Das 1 , Md. Golam Rosul 2 , Mona Zebarjadi 1, 3
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-03-31 , DOI: 10.1021/acs.jpcc.2c09137
Sree Sourav Das 1 , Md. Golam Rosul 2 , Mona Zebarjadi 1, 3
Affiliation
|
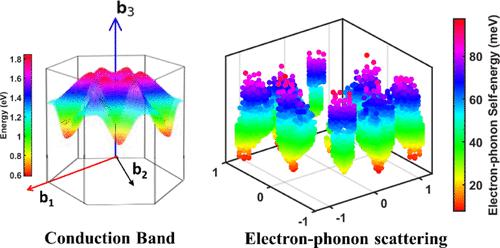
我们提出了一个第一性原理框架来研究 SnSe 2的电子特性,一种潜在的良好层状热电材料。我们使用密度泛函理论和玻尔兹曼输运方程在弛豫时间近似下的解,包括电子-声子和电离杂质相互作用来计算热电功率因数,其中使用 PERTUBO 包计算电子-声子散射,改进的 Brooks-Herring 方法是用于模拟电离杂质散射。我们研究了在包含和不包含范德瓦尔斯相互作用的情况下不同载流子浓度下的温度依赖性传输特性。包含范德瓦尔斯相互作用会增加电子-声子散射,但总弛豫时间主要由高浓度水平的电离杂质散射决定。2作为温度和载流子浓度的函数。优化的功率因数乘以温度发生在载流子浓度为 4 × 10 19 cm –3时,并在面内方向的 523 K 和 0.185 W m –1 K – 处达到最大值 0.49 W m –1 K –1 1在横向方向上 700 K。最后,使用 Callaway 的模型评估晶格热导率。我们的优化模型显示 950 K 时的最高ZT值为 1.1,这源于 SnSe 2层的超低热导率。

"点击查看英文标题和摘要"
更新日期:2023-03-31
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号