Theoretical Chemistry Accounts ( IF 1.6 ) Pub Date : 2023-03-30 , DOI: 10.1007/s00214-023-02972-3 Moulay Driss Mellaoui , Nivedita Acharjee , Abdallah Imjjad , Jamal Koubachi , Abdellatif El Hammadi , Hassan Bourzi , Souad El Issami , Hanane Zejli , Majdi Hochlaf , Khalid Abbiche
|
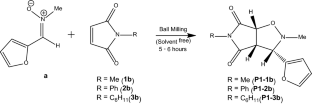
The [3 + 2] cycloaddition (32CA) reactions of N-methyl-C-(2-furyl) nitrone with maleimide derivatives have been studied in gas phase, ethanol and acetonitrile within the Molecular Electron Density Theory (MEDT) framework at the B3LYP-D3/6-31G(d) level. Topological analysis allows classifying the nitrone as a zwitterionic (zw-) three-atom component (TAC) associated with high energy barrier. Interestingly, the global electron density transfer (GEDT) between 0.10 and 0.13 e predicts low polar character of forward electron density transfer (FEDF) type with the electronic flux from the nitrone to the maleimides, resulting in decreased activation parameters relative to that of the nitrone cycloadditions with simple alkenes. These 32CA reactions follow a one-step mechanism under kinetic control with highly asynchronous bond formation, and no new covalent bonds are formed at the TSs. The predicted exo-selectivity agrees well with the experimental findings. Inclusion of solvent effects increases the activation energy, in particular along the endo pathway. The activation enthalpies are between 5.58 and 7.68 kcal/mol in gas phase and between 6.61 and 11.10 kcal/mol in ethanol and acetonitrile. Also, the influence of temperature was investigated at 289.15 K, 298.15 K and 393.15 K. Non-covalent interactions (NCIs) were identified at the interactions regions of the TSs from the topological analysis of the AIM (atoms-in-molecules) and characterized using Independent Gradient Model (IGM) analysis.
中文翻译:
从分子电子密度理论角度揭示N-甲基-C-(2-呋喃基)硝酮与马来酰亚胺衍生物的[3+2]环加成反应的机理、选择性、溶剂和温度影响
在 B3LYP 的分子电子密度理论 (MEDT) 框架内,在气相、乙醇和乙腈中研究了 N -甲基-C-(2-呋喃基)硝酮与马来酰亚胺衍生物的 [3 + 2] 环加成 (32CA )反应-D3/6-31G(d) 级。拓扑分析允许将硝酮分类为两性离子 ( zw-) three-atom component (TAC) associated with high energy barrier. Interestingly, the global electron density transfer (GEDT) between 0.10 and 0.13 e predicts low polar character of forward electron density transfer (FEDF) type with the electronic flux from the nitrone to the maleimides, resulting in decreased activation parameters relative to that of the nitrone cycloadditions with simple alkenes. These 32CA reactions follow a one-step mechanism under kinetic control with highly asynchronous bond formation, and no new covalent bonds are formed at the TSs. The predicted exo-selectivity agrees well with the experimental findings. Inclusion of solvent effects increases the activation energy, in particular along the endo途径。活化焓在气相中介于 5.58 和 7.68 kcal/mol 之间,在乙醇和乙腈中介于 6.61 和 11.10 kcal/mol 之间。此外,还在 289.15 K、298.15 K 和 393.15 K 处研究了温度的影响。通过 AIM(分子中的原子)的拓扑分析,在 TS 的相互作用区域识别出非共价相互作用 (NCI),并对其进行了表征使用独立梯度模型 (IGM) 分析。








































 京公网安备 11010802027423号
京公网安备 11010802027423号