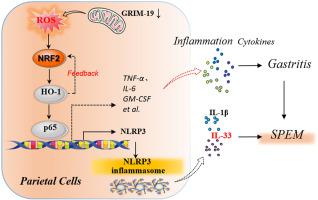
表达解痉多肽的化生 (SPEM) 作为肠化生 (IM) 的癌前前体,在慢性萎缩性胃炎 (CAG) 和胃癌 (GC) 的发展中起着关键作用。然而,导致 SPEM 发病机制的致病靶标仍然知之甚少。与维甲酸-干扰素诱导的死亡率相关的基因 19 ( GRIM-19 ) 是线粒体呼吸链复合体 I 的一个重要亚基,随着人类 CAG 的恶性转化而逐渐丢失,关于 GRIM-19 丢失与 GRIM-19 丢失之间的潜在联系知之甚少和 CAG 发病机制。在这里,我们表明较低的GRIM-19与 CAG 病变中较高的 NF-кB RelA/p65 和 NLR 家族含 pyrin 结构域 3 (NLRP3) 水平相关。在功能上,GRIM-19缺陷不能在体外驱动人类 GES-1 细胞直接分化为 IM 或 SPEM 样细胞谱系,而壁细胞 (PC) 特异性GRIM-19敲除会干扰胃腺分化并促进自发性胃炎和 SPEM 发病机制,而无需小鼠的肠道特征。从机制上讲,GRIM-19丢失导致慢性粘膜损伤和异常 NRF2(核因子红细胞 2 相关因子 2)- HO-1(血红素加氧酶-1)通过活性氧(ROS)介导的氧化应激激活,导致异常 NF -通过 IKK/IкB 合作伙伴诱导 p65 核易位激活 kB,而 NRF2-HO-1 激活有助于GRIM-19通过正反馈 NRF2-HO-1 回路损失驱动的 NF-кB 激活。此外,GRIM-19丢失不会导致明显的 PC 丢失,但会通过 ROS-NRF2-HO-1-NF-кB 轴触发 PC 中的 NLRP3 炎性体激活,导致 NLRP3 依赖性 IL-33 表达,这是 SPEM 形成的关键介质. 此外,NLRP3 抑制剂 MCC950 的腹膜内给药可显着减弱体内GRIM -19丢失驱动的胃炎和 SPEM 。我们的研究表明,线粒体GRIM-19可能是 SPEM 发病机制的潜在致病靶点,其缺陷通过 ROS-NRF2-HO-1-NF-кB 轴通过 NLRP3/IL-33 途径促进 SPEM。这一发现不仅提供了GRIM-19之间的因果联系丢失和 SPEM 发病机制,但为肠道 GC 的早期预防提供了潜在的治疗策略。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Mitochondrial GRIM-19 loss in parietal cells promotes spasmolytic polypeptide-expressing metaplasia through NLR family pyrin domain-containing 3 (NLRP3)-mediated IL-33 activation via a reactive oxygen species (ROS) -NRF2- Heme oxygenase-1(HO-1)–NF–кB axis
Spasmolytic polypeptide-expressing metaplasia (SPEM), as a pre-neoplastic precursor of intestinal metaplasia (IM), plays critical roles in the development of chronic atrophic gastritis (CAG) and gastric cancer (GC). However, the pathogenetic targets responsible for the SPEM pathogenesis remain poorly understood. Gene associated with retinoid–IFN–induced mortality 19 (GRIM-19), an essential subunit of the mitochondrial respiratory chain complex I, was progressively lost along with malignant transformation of human CAG, little is known about the potential link between GRIM-19 loss and CAG pathogenesis. Here, we show that lower GRIM-19 is associated with higher NF-кB RelA/p65 and NLR family pyrin domain-containing 3 (NLRP3) levels in CAG lesions. Functionally, GRIM-19 deficiency fails to drive direct differentiation of human GES-1 cells into IM or SPEM-like cell lineages in vitro, whereas parietal cells (PCs)-specific GRIM-19 knockout disturbs gastric glandular differentiation and promotes spontaneous gastritis and SPEM pathogenesis without intestinal characteristics in mice. Mechanistically, GRIM-19 loss causes chronic mucosal injury and aberrant NRF2 (Nuclear factor erythroid 2-related factor 2)- HO-1 (Heme oxygenase-1) activation via reactive oxygen species (ROS)-mediated oxidative stress, resulting in aberrant NF-кB activation by inducing p65 nuclear translocation via an IKK/IкB partner, while NRF2-HO-1 activation contributes to GRIM-19 loss-driven NF-кB activation via a positive feedback NRF2-HO-1 loop. Furthermore, GRIM-19 loss did not cause obvious PCs loss but triggers NLRP3 inflammasome activation in PCs via a ROS-NRF2-HO-1-NF-кB axis, leading to NLRP3-dependent IL-33 expression, a key mediator for SPEM formation. Moreover, intraperitoneal administration of NLRP3 inhibitor MCC950 drastically attenuates GRIM-19 loss-driven gastritis and SPEM in vivo. Our study suggests that mitochondrial GRIM-19 maybe a potential pathogenetic target for the SPEM pathogenesis, and its deficiency promotes SPEM through NLRP3/IL-33 pathway via a ROS-NRF2-HO-1-NF-кB axis. This finding not only provides a causal link between GRIM-19 loss and SPEM pathogenesis, but offers potential therapeutic strategies for the early prevention of intestinal GC.





























 京公网安备 11010802027423号
京公网安备 11010802027423号