Life Sciences ( IF 5.2 ) Pub Date : 2023-03-21 , DOI: 10.1016/j.lfs.2023.121608 Xiaolin Sun 1 , Ning Huang 1 , Peng Li 1 , Xinyi Dong 1 , Jiahong Yang 1 , Xuemei Zhang 1 , Wei-Xing Zong 2 , Shenglan Gao 3 , Hong Xin 1
|
Aims
This study aims to verify the molecular mechanism that Tripartite motif containing 21 (TRIM21) promotes ubiquitination degradation of glutathione peroxidase 4 (GPX4) by regulating ferroptosis, and to discuss the feasibility of TRIM21 as a new therapeutic target for acute kidney injury (AKI).
Materials and methods
Ischemia-reperfusion (I/R)-AKI model was constructed using Trim21+/+ and Trim21−/− mice, and the expression of markers associated with kidney injury and ferroptosis were evaluated. HK-2 cells were treated by RSL3 and Erastin, and a hypoxia/reoxygenation (H/R) model was constructed to simulate I/R injury in vivo.
Key findings
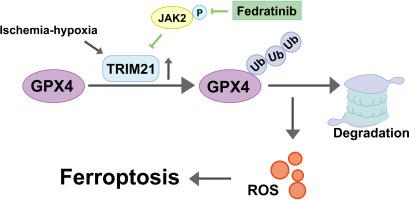
In vivo, TRIM21 is highly expressed in I/R kidney tissues. Loss of TRIM21 alleviated I/R-AKI and improved renal function. The upregulation of GPX4, a key ferroptosis regulator, and the mild mitochondrial damage suggested that loss of TRIM21 had a negative regulation of ferroptosis. In vitro, TRIM21 was highly expressed in H/R models, and overexpression of TRIM21 in HK-2 cells increased ROS production, promoted intracellular iron accumulation, and boosted cellular sensitivity to RSL3 and Erastin. Mechanistically, we confirmed that GPX4 is a substrate of TRIM21 and can be degraded by TRIM21-mediated ubiquitination, suggesting that inhibiting TRIM21 attenuates ferroptosis. A JAK2 inhibitor Fedratinib downregulated TRIM21 expression and reduced damage both in vivo and in vitro, which is correlated with the upregulation of GPX4.
Significance
Our study showed that loss of TRIM21 could alleviate ferroptosis induced by I/R, revealed the mechanism of ubiquitination degradation of GPX4 by TRIM21 and suggested TRIM21 is a potential target for the treatment of AKI.
中文翻译:
TRIM21 泛素化 GPX4 并促进铁死亡以加重缺血/再灌注诱导的急性肾损伤
目标
本研究旨在验证包含 21 的三联基序 (TRIM21) 通过调节铁死亡促进谷胱甘肽过氧化物酶 4 (GPX4) 泛素化降解的分子机制,并探讨 TRIM21 作为急性肾损伤 (AKI) 新治疗靶点的可行性。
材料和方法
使用 Trim21+/+ 和 Trim21−/− 小鼠构建缺血再灌注 (I/R)-AKI 模型,并评估与肾损伤和铁死亡相关的标志物的表达。用 RSL3 和 Eastin 处理 HK-2 细胞,构建缺氧/复氧 (H/R) 模型模拟体内 I/R 损伤。
主要发现
在体内,TRIM21 在 I/R 肾组织中高表达。TRIM21 的缺失减轻了 I/R-AKI 并改善了肾功能。关键铁死亡调节因子 GPX4 的上调和轻度线粒体损伤表明 TRIM21 的缺失对铁死亡具有负调节作用。在体外,TRIM21 在 H/R 模型中高表达,在 HK-2 细胞中过表达 TRIM21 增加了 ROS 的产生,促进了细胞内铁的积累,并提高了细胞对 RSL3 和 Eastin 的敏感性。从机制上讲,我们证实 GPX4 是 TRIM21 的底物,可以被 TRIM21 介导的泛素化降解,表明抑制 TRIM21 会减轻铁死亡。JAK2 抑制剂 Fedratinib 下调 TRIM21 表达并减少体内和体外损伤,这与 GPX4 的上调相关。
意义
我们的研究表明,TRIM21 的缺失可以减轻 I/R 诱导的铁死亡,揭示了 TRIM21 对 GPX4 泛素化降解的机制,并表明 TRIM21 是治疗 AKI 的潜在靶点。






























 京公网安备 11010802027423号
京公网安备 11010802027423号