Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2023-03-11 , DOI: 10.1016/j.molstruc.2023.135320 K.M Chandini , T. N Lohith , S Shamanth , M. A Sridhar , K Mantelingu , N. K Lokanath
|
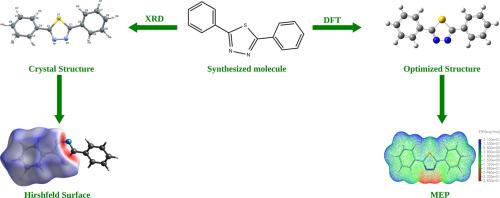
A novel thiadiazole derivative, 2,5-diphenyl-1,3,4-thiadiazole has been synthesized and characterized by elemental analyses 1HNMR and 13CNMR . The crystal structure of the title compound was determined by the single crystal X-ray diffraction study. The compound crystallizes in the monoclinic crystal system with space group C 2/c and cell parameters a = 27.478(11) Å, b = 5.761(2) Å, c = 7.353(3) Å, β = 106.6°, and Z = 4. The R factor converges to 0.0360. The 3D molecular energy frameworks were constructed using the interaction energies. Density Functional Theory (DFT) calculations were performed to optimize the structural coordinates and the results were correlated with the XRD results. The HOMO-LUMO energy gap, and other electronic parameters of the title molecule were calculated.
中文翻译:
2,5-二苯基-1,3,4-噻二唑的合成、结构解析、能量框架和 DFT 计算
合成了一种新型噻二唑衍生物 2,5-二苯基-1,3,4-噻二唑,并通过元素分析1 HNMR 和13 CNMR 对其进行了表征。标题化合物的晶体结构通过单晶 X 射线衍射研究确定。该化合物在单斜晶系中结晶,空间群为C 2 /c,晶胞参数a = 27.478(11) Å, b = 5.761(2) Å, c = 7.353(3) Å, β = 106.6°, Z = 4. R因子收敛到 0.0360 。3D 分子能量框架是使用相互作用能量构建的。进行密度泛函理论 (DFT) 计算以优化结构坐标,并将结果与 XRD 结果相关联。计算了标题分子的 HOMO-LUMO 能隙和其他电子参数。





























 京公网安备 11010802027423号
京公网安备 11010802027423号