当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Study on the Catalytic CO2 Hydrogenation over the MOF-808-Encapsulated Single-Atom Metal Catalysts
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-02-21 , DOI: 10.1021/acs.jpcc.2c08006
Jinlu Liu 1 , Wenjuan Xue 1 , Weiwei Zhang 1 , Donghai Mei 2
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-02-21 , DOI: 10.1021/acs.jpcc.2c08006
Jinlu Liu 1 , Wenjuan Xue 1 , Weiwei Zhang 1 , Donghai Mei 2
Affiliation
|
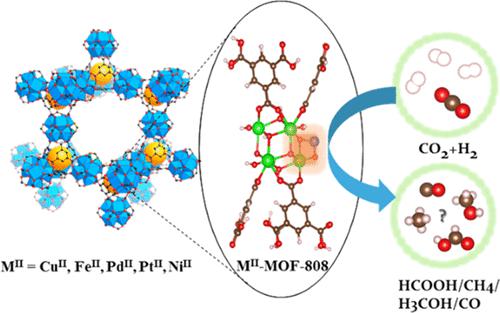
The search for new catalytic agents for reducing excess CO2 in the atmosphere is a challenging but essential task. Due to the well-defined porous structures and unique physicochemical properties, metal–organic frameworks (MOFs) have been regarded as one of the promising materials in the catalytic conversion of CO2 into valuable platform chemicals. In particular, introducing the second metal (M) atom to form the MII–O–Zr4+ single-atom metal sites on the Zr nodes of MOF-808 would further greatly improve the catalytic performance. Herein, CO2 hydrogenation reaction mechanisms and kinetics over a series of MOF-808-encapsulated single-atom metal catalysts, i.e., MII–MOF-808 (MII = CuII, FeII, PtII, NiII, and PdII), were systematically studied using density functional theory calculations. First, it has been found that the stability for the encapsulation of a divalent metal ion follows the trend of PtII > NiII > PdII > CuII > FeII, while they all possess moderate anchoring stability on the MOF-808 with the Gibbs replacement energies ranging from −233.7 to −310.3 kcal/mol. Two plausible CO2 hydrogenation pathways on CuII–MOF-808 catalysts, i.e., formate and carboxyl routes, were studied. The formate route is more favorable, in which the H2COOH*-to-H2CO* step is kinetically the most relevant step over CuII–MOF-808. Using the energetic span model, the relative turnover frequencies of CO2 hydrogenation to various C1 products over MII–MOF-808 were calculated. The CuII–MOF-808 catalyst is found to be the most active catalyst among five MII–MOF-808 catalysts.
中文翻译:

MOF-808包封单原子金属催化剂催化CO2加氢的理论研究
寻找新的催化剂来减少大气中过量的 CO 2是一项具有挑战性但必不可少的任务。由于明确定义的多孔结构和独特的物理化学性质,金属有机骨架 (MOF) 被认为是将 CO 2催化转化为有价值的平台化学品的有前途的材料之一。特别地,引入第二个金属(M)原子在 MOF-808 的 Zr 节点上形成 M II –O–Zr 4+单原子金属位点将进一步大大提高催化性能。在此,CO 2加氢反应机制和动力学在一系列 MOF-808 封装的单原子金属催化剂上,即 M II –MOF-808 (MII = Cu II、Fe II、Pt II、Ni II和 Pd II ),使用密度泛函理论计算进行了系统研究。首先,已经发现二价金属离子的包封稳定性遵循 Pt II > Ni II > Pd II > Cu II > Fe II的趋势,而它们在 MOF-808 上都具有中等的锚定稳定性,吉布斯置换能范围为 -233.7 至 -310.3 kcal/mol。Cu II上两种可能的 CO 2加氢途径– 研究了 MOF-808 催化剂,即甲酸酯和羧基路线。甲酸盐路线更有利,其中 H 2 COOH*-to-H 2 CO* 步骤在动力学上是与 Cu II –MOF-808最相关的步骤。使用能量跨度模型,计算了M II –MOF-808 上 CO 2加氢生成各种 C1 产物的相对周转频率。发现Cu II –MOF-808 催化剂是五种 M II –MOF-808 催化剂中活性最高的催化剂。
更新日期:2023-02-21
中文翻译:
MOF-808包封单原子金属催化剂催化CO2加氢的理论研究
寻找新的催化剂来减少大气中过量的 CO 2是一项具有挑战性但必不可少的任务。由于明确定义的多孔结构和独特的物理化学性质,金属有机骨架 (MOF) 被认为是将 CO 2催化转化为有价值的平台化学品的有前途的材料之一。特别地,引入第二个金属(M)原子在 MOF-808 的 Zr 节点上形成 M II –O–Zr 4+单原子金属位点将进一步大大提高催化性能。在此,CO 2加氢反应机制和动力学在一系列 MOF-808 封装的单原子金属催化剂上,即 M II –MOF-808 (MII = Cu II、Fe II、Pt II、Ni II和 Pd II ),使用密度泛函理论计算进行了系统研究。首先,已经发现二价金属离子的包封稳定性遵循 Pt II > Ni II > Pd II > Cu II > Fe II的趋势,而它们在 MOF-808 上都具有中等的锚定稳定性,吉布斯置换能范围为 -233.7 至 -310.3 kcal/mol。Cu II上两种可能的 CO 2加氢途径– 研究了 MOF-808 催化剂,即甲酸酯和羧基路线。甲酸盐路线更有利,其中 H 2 COOH*-to-H 2 CO* 步骤在动力学上是与 Cu II –MOF-808最相关的步骤。使用能量跨度模型,计算了M II –MOF-808 上 CO 2加氢生成各种 C1 产物的相对周转频率。发现Cu II –MOF-808 催化剂是五种 M II –MOF-808 催化剂中活性最高的催化剂。






































 京公网安备 11010802027423号
京公网安备 11010802027423号