客观的
线粒体丙酮酸载体 (MPC) 已成为治疗胰岛素抵抗、2 型糖尿病和非酒精性脂肪性肝炎 (NASH) 的治疗靶点。我们评估了 MPC 抑制剂 (MPCi) 是否可以纠正支链氨基酸 (BCAA) 分解代谢的损伤,这种损伤可预测糖尿病和 NASH 的发生。
方法
对 NASH 和 2 型糖尿病患者的循环 BCAA 浓度进行了测量,这些患者参加了最近一项随机、安慰剂对照的 IIB 期临床试验,以测试 MPCi MSDC-0602K (EMMINENCE;NCT02784444) 的有效性和安全性。在这项为期 52 周的试验中,患者被随机分配接受安慰剂 (n = 94) 或 250 mg MSDC-0602K (n = 101)。使用人肝癌细胞系和小鼠原代肝细胞在体外测试各种MPCi对BCAA分解代谢的直接影响。最后,我们研究了肝细胞特异性删除 MPC2 如何影响肥胖小鼠肝脏中的 BCAA 代谢以及 MSDC-0602K 对 Zucker 糖尿病脂肪 (ZDF) 大鼠的治疗。
结果
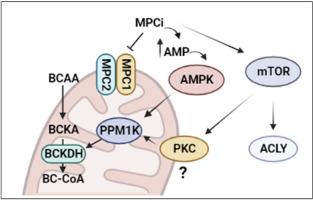
在 NASH 患者中,MSDC-0602K 治疗可显着改善胰岛素敏感性和糖尿病,与基线相比,血浆支链氨基酸浓度降低,而安慰剂则没有效果。 BCAA 分解代谢的限速酶是线粒体支链酮酸脱氢酶 (BCKDH),它会因磷酸化而失活。在多种人肝癌细胞系中,MPCi 显着降低 BCKDH 磷酸化并刺激支链酮酸分解代谢;需要 BCKDH 磷酸酶 PPM1K 的作用。从机制上讲,MPCi 的作用与能量感应 AMP 依赖性蛋白激酶 (AMPK) 的激活和体外雷帕霉素 (mTOR) 激酶信号级联的机制靶标有关。与野生型对照相比,肥胖、肝细胞特异性 MPC2 敲除 (LS- Mpc2 −/−) 小鼠的肝脏中 BCKDH 磷酸化降低,同时体内 mTOR 信号激活。最后,虽然 MSDC-0602K 治疗改善了 ZDF 大鼠的葡萄糖稳态并增加了一些 BCAA 代谢物的浓度,但它并没有降低血浆 BCAA 浓度。
结论
这些数据证明了线粒体丙酮酸和 BCAA 代谢之间的新交互作用,并表明 MPC 抑制通过激活 mTOR 轴导致血浆 BCAA 浓度降低和 BCKDH 磷酸化。然而,MPCi 对葡萄糖稳态的影响可能与其对 BCAA 浓度的影响是分开的。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Mitochondrial pyruvate carrier inhibition initiates metabolic crosstalk to stimulate branched chain amino acid catabolism
Objective
The mitochondrial pyruvate carrier (MPC) has emerged as a therapeutic target for treating insulin resistance, type 2 diabetes, and nonalcoholic steatohepatitis (NASH). We evaluated whether MPC inhibitors (MPCi) might correct impairments in branched chain amino acid (BCAA) catabolism, which are predictive of developing diabetes and NASH.
Methods
Circulating BCAA concentrations were measured in people with NASH and type 2 diabetes, who participated in a recent randomized, placebo-controlled Phase IIB clinical trial to test the efficacy and safety of the MPCi MSDC-0602K (EMMINENCE; NCT02784444). In this 52-week trial, patients were randomly assigned to placebo (n = 94) or 250 mg MSDC-0602K (n = 101). Human hepatoma cell lines and mouse primary hepatocytes were used to test the direct effects of various MPCi on BCAA catabolism in vitro. Lastly, we investigated how hepatocyte-specific deletion of MPC2 affects BCAA metabolism in the liver of obese mice and MSDC-0602K treatment of Zucker diabetic fatty (ZDF) rats.
Results
In patients with NASH, MSDC-0602K treatment, which led to marked improvements in insulin sensitivity and diabetes, had decreased plasma concentrations of BCAAs compared to baseline while placebo had no effect. The rate-limiting enzyme in BCAA catabolism is the mitochondrial branched chain ketoacid dehydrogenase (BCKDH), which is deactivated by phosphorylation. In multiple human hepatoma cell lines, MPCi markedly reduced BCKDH phosphorylation and stimulated branched chain keto acid catabolism; an effect that required the BCKDH phosphatase PPM1K. Mechanistically, the effects of MPCi were linked to activation of the energy sensing AMP-dependent protein kinase (AMPK) and mechanistic target of rapamycin (mTOR) kinase signaling cascades in vitro. BCKDH phosphorylation was reduced in liver of obese, hepatocyte-specific MPC2 knockout (LS-Mpc2−/−) mice compared to wild-type controls concomitant with activation of mTOR signaling in vivo. Finally, while MSDC-0602K treatment improved glucose homeostasis and increased the concentrations of some BCAA metabolites in ZDF rats, it did not lower plasma BCAA concentrations.
Conclusions
These data demonstrate novel cross talk between mitochondrial pyruvate and BCAA metabolism and suggest that MPC inhibition leads to lower plasma BCAA concentrations and BCKDH phosphorylation by activating the mTOR axis. However, the effects of MPCi on glucose homeostasis may be separable from its effects on BCAA concentrations.




















































 京公网安备 11010802027423号
京公网安备 11010802027423号