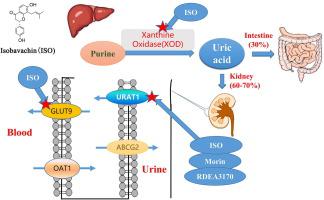
之前,我们发现了一种新型天然支架化合物异巴伐他汀 (4', 7-dihydroxy-8-prenylflavanone),它是一种基于美德筛选方法的形状和结构有效的 URAT1 抑制剂。在本研究中,进一步对异巴巴星的降尿酸机制、药代动力学和毒性进行了研究。Isobavachin 以0.24 ± 0.06 μM 的IC 50值抑制 URAT1,并且 URAT1 的残基 S35、F365、I481 和 R477 有助于对 isobavachin 的高亲和力。Isobavachin 还抑制葡萄糖转运蛋白 9 (GLUT9),这是另一种关键的尿酸盐再吸收转运蛋白,IC 为50值为 1.12 ± 0.26 μM。分子对接和 MMGBSA 结果表明,isobavachin 可能与尿酸竞争残基 R171、L75 和 N333,从而抑制 GLUT9 的尿酸转运。Isobavachin 微弱抑制尿酸盐分泌转运蛋白 OAT1,IC 50值为 4.38 ± 1.27 μM,OAT3 IC 50为 3.64 ± 0.62 μM,ABCG2 IC 50为 10.45 ± 2.17 μM。Isobavachin 还在体外抑制黄嘌呤氧化酶 (XOD) 活性,IC 50值为 14.43 ± 3.56 μM,并在 5–20 mg/kg体内抑制肝脏 XOD 活性. 对接和 MMGBSA 分析表明,isobavachin 可能与 XOD 的 Mo-Pt 催化中心结合,从而抑制尿酸的产生。在体内,与阳性药物桑葚素 (20 mg/kg) 和 RDEA3170 (10 mg/kg) 相比,isobavachin 在 5–20 mg/kg 时表现出强大的降尿酸和排尿酸作用。安全性评估表明,异巴巴星是安全的,没有明显的毒性。如 MTT 测定所示,Isobavachin 在 HK2 细胞中几乎没有细胞毒性。体内,用 50 mg/kg isobavachin 治疗 14 天后,isobavachin 几乎没有肾毒性,如血清 CR/BUN 水平所示,而无肝毒性,如 ALT/AST 水平所示。进一步的HE检查也表明异巴伐他汀没有明显的肾/肝损伤。在 SD 大鼠中进行的药代动力学研究表明,异巴巴星的生物利用度较低 (12.84 ± 5.13 %),但半衰期较长 (7.04 ± 2.68 h) 以维持连续的血浆浓度。总的来说,这些结果表明,异巴巴星作为具有新作用机制的候选抗高尿酸血症药物值得进一步研究:选择性尿酸盐重吸收抑制剂 (URAT1/GLUT9),对 XOD 具有中度抑制作用。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Pharmacological evaluation of a novel skeleton compound isobavachin (4′,7-dihydroxy-8-prenylflavanone) as a hypouricemic agent: Dual actions of URAT1/GLUT9 and xanthine oxidase inhibitory activity
Previously we discovered a novel natural scaffold compound, isobavachin (4′, 7-dihydroxy-8-prenylflavanone), as a potent URAT1 inhibitor by shape and structure based on a virtue screening approach. In this study, further urate-lowering mechanism, pharmacokinetics and toxicities of isobavachin were conducted. Isobavachin inhibited URAT1 with an IC50 value of 0.24 ± 0.06 μM, and residues S35, F365, I481 and R477 of URAT1 contributed to high affinity for isobavachin. Isobavachin also inhibited glucose transporter 9 (GLUT9), another pivotal urate reabsorption transporter, with an IC50 value of 1.12 ± 0.26 μM. Molecular docking and MMGBSA results indicated that isobavachin might compete residues R171, L75 and N333 with uric acid, which leads to inhibition of uric acid transport of GLUT9. Isobavachin weakly inhibited urate secretion transporters OAT1 with an IC50 value of 4.38 ± 1.27 μM, OAT3 with an IC50 of 3.64 ± 0.62 μM, and ABCG2 with an IC50 of 10.45 ± 2.17 μM. Isobavachin also inhibited xanthine oxidase (XOD) activity in vitro with an IC50 value of 14.43 ± 3.56 μM, and inhibited the hepatic XOD activities at 5–20 mg/kg in vivo. Docking and MMGBSA analysis indicated that isobavachin might bind to the Mo-Pt catalyze center of XOD, which leads to inhibition of uric acid production. In vivo, isobavachin exhibited powerful urate-lowering and uricosuric effects at 5–20 mg/kg compared with the positive drugs morin (20 mg/kg) and RDEA3170 (10 mg/kg). Safety assessments revealed that isobavachin was safe and had no obvious toxicities. Isobavachin has little cell toxicity in HK2 cells as indicated by the MTT assay. In vivo, after treatment with 50 mg/kg isobavachin for 14 days, isobavachin had little renal toxicity, as revealed by serum CR/BUN levels, and no hepatotoxicity as revealed by ALT/AST levels. Further HE examination also suggests that isobavachin has no obvious kidney/liver damage. A pharmacokinetic study in SD rats indicated isobavachin had lower bioavailability (12.84 ± 5.13 %) but long half-time (7.04 ± 2.68 h) to maintain a continuous plasma concentration. Collectively, these results indicate that isobavachin deserves further investigation as a candidate anti-hyperuricemic drug with a novel mechanism of action: selective urate reabsorption inhibitor (URAT1/GLUT9) with a moderate inhibitory effect on XOD.






























 京公网安备 11010802027423号
京公网安备 11010802027423号