当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantum Dynamics and Bi Metal Force Field Parameterization Yielding Significant Antileishmanial Targets
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2023-02-02 , DOI: 10.1021/acs.jcim.2c01100 Naila Zaman 1 , Syed Sikander Azam 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2023-02-02 , DOI: 10.1021/acs.jcim.2c01100 Naila Zaman 1 , Syed Sikander Azam 1
Affiliation
|
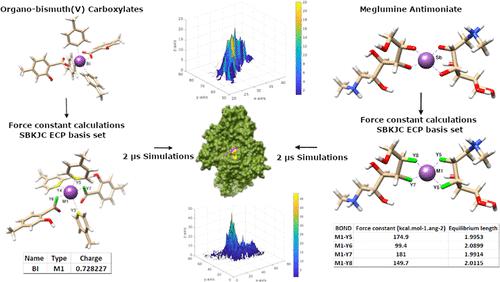
Amid emerging drug resistance to metal inhibitors, high toxicity, and onerous drug delivery procedures, the computational design of alternate formulations encompassing functional metal-containing compounds greatly relies on large-scale atomistic simulations. Simulations particularly with Au(I), Ag, Bi(V), and Sb(V) pose a major challenge to elucidate their molecular mechanism due to the absence of force field parameters. This study thus quantum mechanically derives force field parameters of Bi(V) as an extension of the previous experimental study conducted on heteroleptic triorganobismuth(V) biscarboxylates of type [BiR3(O2CR′)2]. We have modeled two organo-bismuth(V) carboxylates, which are optimized and parameterized along with the famous pentavalent antimonial drug: meglumine antimoniate using quantum mechanics original Seminarian methods with the SBKJC effective core potential (ECP) basis set. Furthermore, molecular dynamics (MD) simulations of bismuth- and antimony-containing compounds in complex with two enzymes, trypanothione synthetase-amidase (TSA) and trypanothione reductase, are performed to target the (T(SH)2) pathway at multiple points. MD simulations provide novel insights into the binding mechanism of TSA and highlight the role of a single residue Arg569 in modulating the ligand dynamics. Moreover, the presence of an ortho group in a ligand is emphasized to facilitate interactions between Arg569 and the active site residue Arg313 for higher inhibitory activity of TSA. This preliminary generation of parameters specific to bismuth validated by simulations in replica will become a preamble of future computational and experimental research work to open avenues for newer and suitable drug targets.
中文翻译:

量子动力学和双金属力场参数化产生重要的反利什曼目标
在新出现的金属抑制剂耐药性、高毒性和繁重的药物输送程序中,包含功能性含金属化合物的替代配方的计算设计在很大程度上依赖于大规模原子模拟。由于缺乏力场参数,特别是 Au(I)、Ag、Bi(V) 和 Sb(V) 的模拟对阐明其分子机制构成了重大挑战。因此,本研究通过量子力学推导了 Bi(V) 的力场参数,作为先前对 [BiR 3 (O 2 CR′) 2 ] 型杂配三有机铋 (V) 双羧酸盐进行的实验研究的延伸。我们使用量子力学原始 Seminarian 方法和 SBKJC 有效核心电位 (ECP) 基础集对两种有机铋 (V) 羧酸盐进行了建模,并与著名的五价锑药物:葡甲胺锑酸盐一起进行了优化和参数化。此外,对含铋和锑的化合物与两种酶——锥硫酮合成酶-酰胺酶(TSA)和锥硫酮还原酶——进行复合物的分子动力学(MD)模拟,以在多个点靶向(T(SH)2)途径。MD 模拟为 TSA 的结合机制提供了新的见解,并强调了单个残基 Arg569 在调节配体动力学中的作用。此外,强调配体中邻位基团的存在,以促进 Arg569 和活性位点残基 Arg313 之间的相互作用,从而实现更高的 TSA 抑制活性。通过复制模拟验证的初步生成的特定于铋的参数将成为未来计算和实验研究工作的序言,为更新和合适的药物靶点开辟途径。
更新日期:2023-02-02
中文翻译:
量子动力学和双金属力场参数化产生重要的反利什曼目标
在新出现的金属抑制剂耐药性、高毒性和繁重的药物输送程序中,包含功能性含金属化合物的替代配方的计算设计在很大程度上依赖于大规模原子模拟。由于缺乏力场参数,特别是 Au(I)、Ag、Bi(V) 和 Sb(V) 的模拟对阐明其分子机制构成了重大挑战。因此,本研究通过量子力学推导了 Bi(V) 的力场参数,作为先前对 [BiR 3 (O 2 CR′) 2 ] 型杂配三有机铋 (V) 双羧酸盐进行的实验研究的延伸。我们使用量子力学原始 Seminarian 方法和 SBKJC 有效核心电位 (ECP) 基础集对两种有机铋 (V) 羧酸盐进行了建模,并与著名的五价锑药物:葡甲胺锑酸盐一起进行了优化和参数化。此外,对含铋和锑的化合物与两种酶——锥硫酮合成酶-酰胺酶(TSA)和锥硫酮还原酶——进行复合物的分子动力学(MD)模拟,以在多个点靶向(T(SH)2)途径。MD 模拟为 TSA 的结合机制提供了新的见解,并强调了单个残基 Arg569 在调节配体动力学中的作用。此外,强调配体中邻位基团的存在,以促进 Arg569 和活性位点残基 Arg313 之间的相互作用,从而实现更高的 TSA 抑制活性。通过复制模拟验证的初步生成的特定于铋的参数将成为未来计算和实验研究工作的序言,为更新和合适的药物靶点开辟途径。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号