Nature Communications ( IF 14.7 ) Pub Date : 2023-02-01 , DOI: 10.1038/s41467-023-36103-0 Long Huang 1 , Marcin Szewczyk 1 , Rajesh Kancherla 2 , Bholanath Maity 2 , Chen Zhu 2 , Luigi Cavallo 2 , Magnus Rueping 2, 3
|
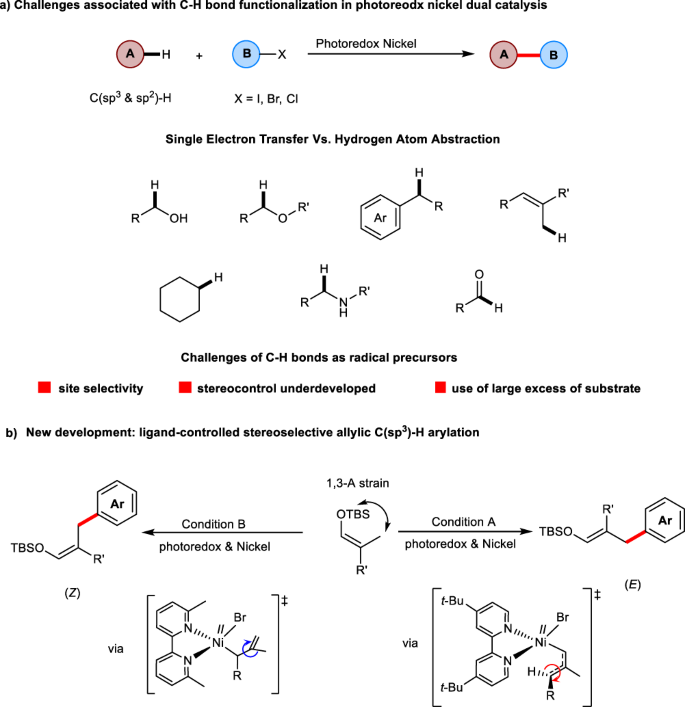
While significant progress has been made in developing selective C-H bond cross-couplings in the field of radical chemistry, the site and stereoselectivity remain a long-standing challenge. Here, we present the successful development of stereodivergent allylic C(sp3)-H bond arylations through a systematic investigation of the direction and degree of stereoselectivity in the cross-coupling process. In contrast to the signature photosensitized geometrical isomerization of alkenes, the catalytic reaction demonstrates the feasibility of switching the C-C double bond stereoselectivity by means of ligand control as well as steric and electronic effects. Computational studies explain the stereochemical outcome and indicate that excitation of a Ni-allyl complex from singlet to a triplet state results in a spontaneous change of the allyl group coordination and that the subsequent isomerization can be directed by the choice of the ligand to achieve E/Z selectivity.
中文翻译:
通过镍和光氧化还原催化调节烯丙基 C(sp3)-H 键芳基化的立体选择性
虽然自由基化学领域在开发选择性 CH 键交叉偶联方面取得了重大进展,但其位点和立体选择性仍然是一个长期存在的挑战。在这里,我们介绍了立体发散烯丙基 C(sp 3)-H 键芳基化,通过对交叉偶联过程中立体选择性的方向和程度的系统研究。与烯烃的标志性光敏几何异构化相反,催化反应证明了通过配体控制以及空间和电子效应来切换 CC 双键立体选择性的可行性。计算研究解释了立体化学结果,并表明将 Ni-烯丙基络合物从单线态激发到三线态会导致烯丙基配位的自发变化,并且随后的异构化可以通过配体的选择来实现E / Z选择性。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号