当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
α-SiO2 表面作为 Pd 载体的计算研究
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2023-01-30 , DOI: 10.1039/d2cp04722e
C J Lombard 1 , C G C E van Sittert 1 , J N Mugo 2 , C Perry 3 , D J Willock 4
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2023-01-30 , DOI: 10.1039/d2cp04722e
C J Lombard 1 , C G C E van Sittert 1 , J N Mugo 2 , C Perry 3 , D J Willock 4
Affiliation
|
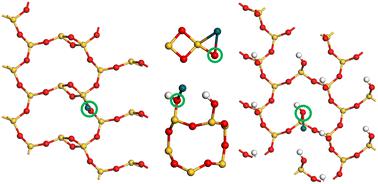
负载型金属催化剂的性能主要取决于活性金属与载体之间的相互作用。典型的例子是负载在二氧化硅上的 Pd,即 Pd/SiO 2,它广泛用于氧化催化。需要广泛的计算模型来描述 Pd 与二氧化硅表面的相互作用,以便可以提出和测试活性位点模型。在这项工作中,我们创建了 SiO 2表面定义明确、可重现的周期性模型,并使用色散校正 DFT 研究了它们与 Pd 的相互作用。我们使用结晶 α-SiO 2作为创建和估计金属在 SiO 2上的吸附特性的有用起点表面,它可以代表更复杂的无定形二氧化硅表面上存在的特定隔离官能团。我们模拟了包含分离的硅氧烷和硅醇官能团的 α-SiO 2 (001)、(100) 和 (101) 表面,并估计了它们对分离的气态 Pd 原子和 fcc Pd 固体吸附 Pd 原子的亲和力。这提供了有关 Pd 可以轻松分散在相关表面上的附加信息。根据我们的模型,我们表征了 α-SiO 2 ( hkl ) 表面的表面能,并计算了每个表面上 Pd 1 /α-SiO 2 ( hkl ) 吸附位点的几何形状。我们估计 Pd 1(g) 更倾向于吸附在紧靠应变四元硅氧烷环或 α-SiO 2 (101) 的邻位硅醇基上。

"点击查看英文标题和摘要"
更新日期:2023-01-30
"点击查看英文标题和摘要"

































 京公网安备 11010802027423号
京公网安备 11010802027423号