Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2023-01-28 , DOI: 10.1016/j.molstruc.2023.135044 Matta Manikanttha , Kolli Deepti , Mandava Bhuvan Tej , Mandava Bhagya Tej , A. Gopi Reddy , Ravikumar Kapavarapu , Deepak Kumar Barange , M.V. Basaveswara Rao , Manojit Pal
|
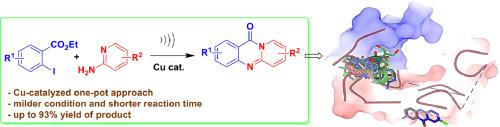
The in silico evaluation of 11H-pyrido[2,1-b]quinazolin-11-one derivatives against SARS-CoV-2 RdRp was undertaken based on the reports on antiviral activities of this class of compounds in addition to the promising interactions of the antiviral drug penciclovir as well as quinazoline derivatives with SARS-CoV-2 RdRp in silico. The target compounds were prepared via an Ullmann–Goldberg type coupling followed by the subsequent cyclization (involving amidation) in a single pot. The methodology involved a CuI-catalyzed reaction of 2-iodobenzoate ester with 2-aminopyridine or quinolin-2-amine or thiazol-2-amine under ultrasound to give the expected products in acceptable (51–93%) yields. The molecular interactions of the synthesized 11H-pyrido[2,1-b]quinazolin-11-one derivatives with the SARS-CoV-2 RdRp (PDB: 7AAP) were evaluated in silico. The study suggested that though none of these compounds showed interactions better than penciclovir but the compound 3a and 3n appeared to be comparable along with 3b seemed to be nearly comparable to favipiravir and remdesivir. The compound 3n with the best binding energy (-79.85 Kcal/mol) participated in the H-bond interactions through its OMe group with THR556 as well as ARG624 and via the N-5 atom with the residue SER682. The in silico studies further suggested that majority of the compounds interacted with the main cavity of active site pocket whereas 3h and 3o that showed relatively lower binding energies (-66.06 and -66.28 Kcal/mol) interacted with the shallow cavity underneath the active site of SARS CoV-2 RdRp. The study also revealed that a OMe group was favourable for interaction with respect to its position in the order C-8 > C-1 > C-2. Further, the presence of a fused quinoline ring was tolerated whereas a fused thiazole ring decreased the interaction significantly. The in silico predictions of pharmacokinetic properties of 3a, 3b and 3n indicated that besides the BBB (Blood Brain Barrier) penetration potential these molecules may show a good overall ADME. Overall, the regioisomers 3a, 3b and 3n have emerged as molecules of possible interest in the context of targeting COVID-19.
中文翻译:
超声辅助 Cu 催化的单锅 Ullmann-Goldberg 型偶联环化:11H-pyrido[2,1-b]quinazolin-11-ones 对 SARS-CoV-2 RdRp 的合成和计算机评价
11 H- pyrido[2,1- b ]quinazolin-11-one 衍生物抗 SARS-CoV-2 RdRp 的计算机评价是根据此类化合物的抗病毒活性报告以及抗病毒药物喷昔洛韦以及喹唑啉衍生物与 SARS-CoV-2 RdRp in silico。目标化合物通过Ullmann-Goldberg 型偶联,随后在单个罐中进行环化(涉及酰胺化)。该方法涉及 2-碘苯甲酸酯与 2-氨基吡啶或喹啉-2-胺或噻唑-2-胺在超声下的 CuI 催化反应,以可接受的 (51–93%) 收率生成预期产物。在计算机上评估了合成的 11 H-吡啶并[2,1 - b ]喹唑啉-11-酮衍生物与 SARS-CoV-2 RdRp (PDB: 7AAP)的分子相互作用。该研究表明,尽管这些化合物中没有一种显示出比喷昔洛韦更好的相互作用,但化合物3a和3n似乎与3b具有可比性似乎几乎可以与 favipiravir 和 remdesivir 相媲美。具有最佳结合能(-79.85 Kcal/mol)的化合物3n通过其OMe基团与THR556和ARG624以及通过N-5原子与残基SER682参与氢键相互作用。计算机研究进一步表明,大多数化合物与活性位点口袋的主腔相互作用,而3h和3o显示相对较低的结合能(-66.06 和 -66.28 Kcal/mol)与 SARS CoV-2 RdRp 活性位点下方的浅腔相互作用。该研究还表明,OMe 组有利于就其位置顺序 C-8 > C-1 > C-2 进行交互。此外,可以容忍稠合喹啉环的存在,而稠合噻唑环会显着降低相互作用。3a、3b和3n的药代动力学特性的计算机预测表明,除了 BBB(血脑屏障)渗透潜力外,这些分子可能显示出良好的整体 ADME。总的来说,区域异构体3a、3b和3n已成为在针对 COVID-19 的背景下可能感兴趣的分子。






























 京公网安备 11010802027423号
京公网安备 11010802027423号