Nature Communications ( IF 14.7 ) Pub Date : 2023-01-12 , DOI: 10.1038/s41467-023-35789-6
Meredith L Jenkins 1 , Harish Ranga-Prasad 1 , Matthew A H Parson 1 , Noah J Harris 1 , Manoj K Rathinaswamy 1 , John E Burke 1, 2
|
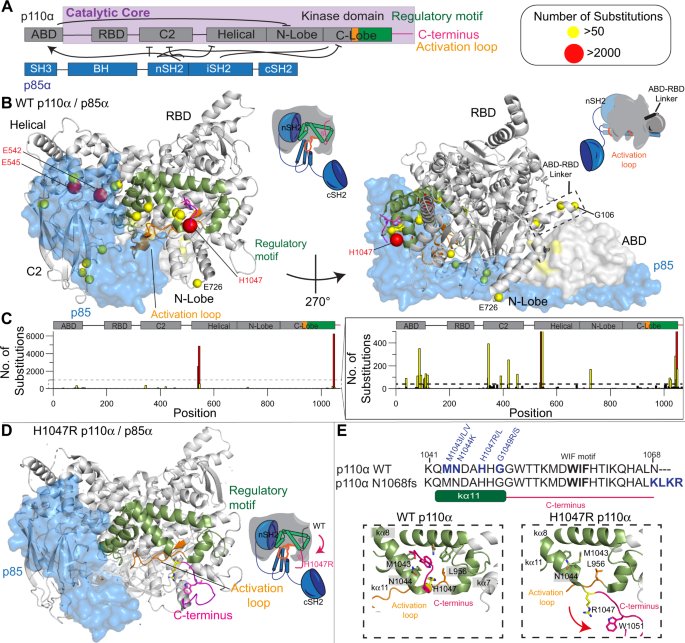
PIK3CA encoding the phosphoinositide 3-kinase (PI3K) p110α catalytic subunit is frequently mutated in cancer, with mutations occurring widely throughout the primary sequence. The full set of mechanisms underlying how PI3Ks are activated by all oncogenic mutations on membranes are unclear. Using a synergy of biochemical assays and hydrogen deuterium exchange mass spectrometry (HDX-MS), we reveal unique regulatory mechanisms underlying PI3K activation. Engagement of p110α on membranes leads to disengagement of the ABD of p110α from the catalytic core, and the C2 domain from the iSH2 domain of the p85 regulatory subunit. PI3K activation also requires reorientation of the p110α C-terminus, with mutations that alter the inhibited conformation of the C-terminus increasing membrane binding. Mutations at the C-terminus (M1043I/L, H1047R, G1049R, and N1068KLKR) activate p110α through distinct mechanisms, with this having important implications for mutant selective inhibitor development. This work reveals unique mechanisms underlying how PI3K is activated by oncogenic mutations, and explains how double mutants can synergistically increase PI3K activity.
中文翻译:
PIK3CA 的致癌突变导致由 ABD、p85 和 C 末端的重新定向驱动的膜募集增加
PIK3CA编码磷酸肌醇 3-激酶 (PI3K) p110α 催化亚基的基因在癌症中经常发生突变,突变在整个一级序列中广泛发生。PI3K 如何被膜上的所有致癌突变激活的全套机制尚不清楚。通过生化分析和氢氘交换质谱 (HDX-MS) 的协同作用,我们揭示了 PI3K 激活的独特调节机制。p110α 在膜上的结合导致 p110α 的 ABD 从催化核心脱离,C2 结构域从 p85 调节亚基的 iSH2 结构域脱离。PI3K 激活还需要重新定位 p110α C 末端,突变会改变 C 末端的抑制构象,从而增加膜结合。C 端突变(M1043I/L、H1047R、G1049R、和 N1068KLKR) 通过不同的机制激活 p110α,这对突变选择性抑制剂的开发具有重要意义。这项工作揭示了 PI3K 如何被致癌突变激活的独特机制,并解释了双突变体如何协同增加 PI3K 活性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号