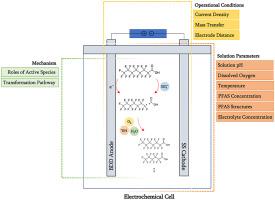
本研究调查了一种用于处理全氟烷基物质和多氟烷基物质 (PFAS) 污染水的电化学方法,研究了不同变量的影响、产生的自由基的贡献以及 PFAS 不同结构的可降解性。从中心复合设计 (CCD) 获得的结果表明传质的重要性,与搅拌速度有关,通过电极的电荷量与电流密度有关,对 PFOA 的分解速率有影响。CCD 告知优化的操作条件,然后我们用它来研究溶液条件的影响。酸性条件、高温和 PFOA 的低初始浓度加速了降解动力学,而 DO 的影响可以忽略不计。电解质浓度的影响取决于 PFOA 的初始浓度。−1 ),当硫酸盐从 0.1% 增加到 10% 时,速率常数从 0.079 ± 0.001 显着增加到 0.259 ± 0.019 min -1 ,这可能是由于 SO 4 •–的产生。然而,在较高的初始 PFOA 剂量 (20 mg L -1 ) 下,速率常数从 0.019 ± 0.001 略微下降至 0.015 ± 0.000 min -1,这可能是由于过量的硫酸盐占据了活性阳极位点。SO 4 •–和• OH 分别在PFOA 的分解和脱氟过程中发挥了重要作用。PFOA 氧化是由一个电子转移到阳极或 SO 4 •–,进行 Kolbe 脱羧反应,其中产生的全氟烷基自由基遵循与• OH、O 2和/或 H 2 O 的三个反应途径。PFAS 电氧化取决于化学结构,其中分解速率常数 (min −1 ) 约为 6:2 FTCA (0.031) > PFOA (0.019) > GenX (0.013) > PFBA (0.008)。链长较短的 PFBA 和带有 –CF 3分支的 GenX 的分解速度比 PFOA 慢。虽然 C-H 键的存在使 6:2 FTCA 容易受到攻击•OH 加速其分解动力学。在所有研究的 PFAS 和天然水中的混合溶液中进行的实验表明,PFAS 和其他水成分(有机物和无机物)的共存对 PFAS 分解效率有不利影响。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Electrochemical degradation of PFOA and its common alternatives: Assessment of key parameters, roles of active species, and transformation pathway
This study investigates an electrochemical approach for the treatment of water polluted with per- and poly-fluoroalkyl substances (PFAS), looking at the impact of different variables, contributions from generated radicals, and degradability of different structures of PFAS. Results obtained from a central composite design (CCD) showed the importance of mass transfer, related to the stirring speed, and the amount of charge passed through the electrodes, related to the current density on decomposition rate of PFOA. The CCD informed optimized operating conditions which we then used to study the impact of solution conditions. Acidic condition, high temperature, and low initial concentration of PFOA accelerated the degradation kinetic, while DO had a negligible effect. The impact of electrolyte concentration depended on the initial concentration of PFOA. At low initial PFOA dosage (0.2 mg L−1), the rate constant increased considerably from 0.079 ± 0.001 to 0.259 ± 0.019 min−1 when sulfate increased from 0.1% to 10%, likely due to the production of SO4•–. However, at higher initial PFOA dosage (20 mg L−1), the rate constant decreased slightly from 0.019 ± 0.001 to 0.015 ± 0.000 min−1, possibly due to the occupation of active anode sites by excess amount of sulfate. SO4•– and •OH played important roles in decomposition and defluorination of PFOA, respectively. PFOA oxidation was initiated by one electron transfer to the anode or SO4•–, undergoing Kolbe decarboxylation where yielded perfluoroalkyl radical followed three reaction pathways with •OH, O2 and/or H2O. PFAS electrooxidation depended on the chemical structures where the decomposition rate constants (min−1) were in the order of 6:2 FTCA (0.031) > PFOA (0.019) > GenX (0.013) > PFBA (0.008). PFBA with a shorter chain length and GenX with –CF3 branching had slower decomposition than PFOA. While presence of C–H bonds makes 6:2 FTCA susceptible to the attack of •OH accelerating its decomposition kinetic. Conducting experiments in mixed solution of all studied PFAS and in natural water showed that the co-presence of PFAS and other water constituents (organic and inorganic matters) had adverse effects on PFAS decomposition efficiency.










































 京公网安备 11010802027423号
京公网安备 11010802027423号