Synthesis ( IF 2.2 ) Pub Date : 2023-01-03 , DOI: 10.1055/a-1984-9689 Nilton Cruz 1 , Amanda Miranda 1 , Henriete Vieira 1 , Markus Kohlhoff 2 , João Mendonça 3 , Marisa Diaz 3 , Gaspar Diaz-Muñoz 1
|
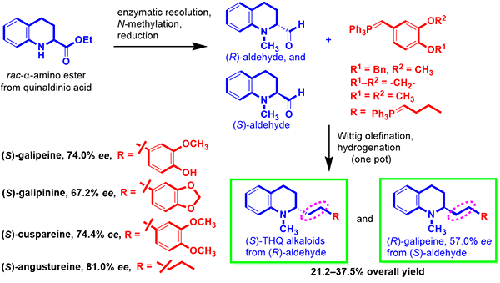
The enantioselective synthesis of the Hancock 1,2,3,4-tetrahydroquinoline alkaloids (S)-galipeine, (S)-cuspareine, (S)-galipinine, and (S)-angustureine and the nonnatural enantiomer (R)-galipeine is described herein. The target compounds were obtained in five steps from a racemic quinaldinic acid derived α-amino ester in overall yields of 21.2% to 37.5%. The synthetic route comprised two key steps: an enzymatic kinetic resolution to control the C-2 stereocenter, affording (R)- and (S)-α-amino esters as key chiral intermediates with 94% and 72% ee, respectively, and Wittig olefination of (R)- and (S)-α-amino aldehyde synthons with the corresponding phosphonium salts using a phase-transfer system (t-BuOH/CH2Cl2), thereby allowing the introduction of alkyl substituents at C-2. Finally, the enantioselective synthesis was concluded with the catalytic hydrogenation of olefinic bonds on the Wittig adducts to furnish the target Hancock alkaloids, including (R)-galipeine, whose synthesis is described here for the first time.
中文翻译:
汉考克生物碱 (S)- 和 (R)-Galipeine、(S)-Cuspareine、(S)-Galipinine 和 (S)-Angustureine 的化学酶对映选择性合成
Hancock 1,2,3,4-四氢喹啉生物碱 ( S )-galipeine、( S )-cuspareine、( S )-galipinine 和 ( S )-angustureine 以及非天然对映异构体 ( R )-galipeine 的对映选择性合成是此处描述。目标化合物由外消旋喹啉甲酸衍生的α-氨基酯经五步制得,总收率为21.2%~37.5%。合成路线包括两个关键步骤:控制 C-2 立体中心的酶促动力学拆分,提供 ( R )- 和 ( S )-α-氨基酯作为关键手性中间体,分别具有 94% 和 72% ee,以及 Wittig ( R )- 和 (S )-α-氨基醛合成子与相应的鏻盐使用相转移系统 ( t -BuOH/CH 2 Cl 2 ),从而允许在 C-2 处引入烷基取代基。最后,对映选择性合成结束于 Wittig 加合物上烯烃键的催化氢化,以提供目标 Hancock 生物碱,包括 ( R )-galipeine,其合成在此首次描述。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号