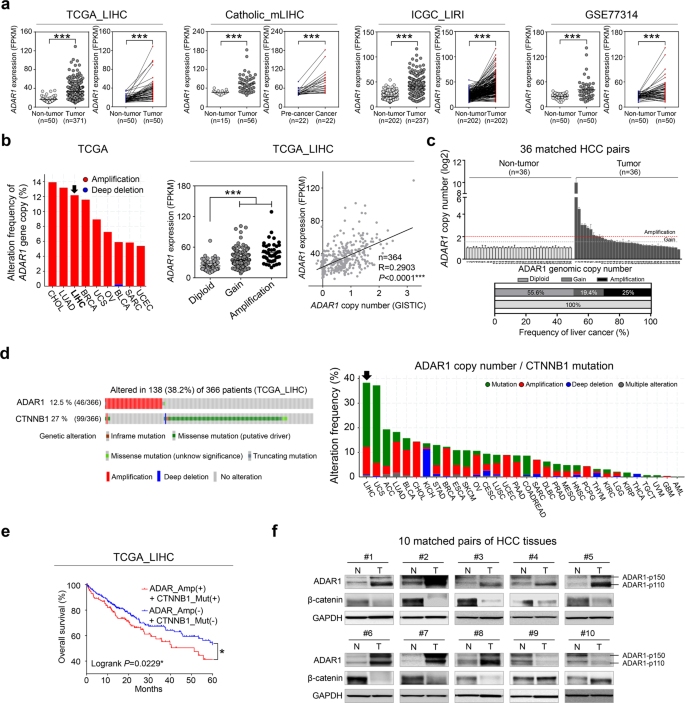
由作用于双链 RNA (ADAR) 的腺苷脱氨酶催化的异常腺苷至肌苷 (A-to-I) RNA 编辑与多种癌症有关,但 microRNA (miRNA) 编辑导致癌症的机制发展在很大程度上是未知的。我们的多阶段肝细胞癌发生转录组数据分析以及公开数据表明,ADAR1是肝癌 RNA 编辑酶家族成员中失调最严重的基因。ADAR1的靶向失活抑制肝癌细胞的体外成瘤。对 RNA 编辑热点和已知 miRNA 编辑频率的综合计算分析表明,miRNA miR-3144-3p 在肝癌进展过程中被 ADAR1 编辑。具体而言,ADAR1 促进了经典 miR-3144-3p 的 A-to-I 编辑,以在肝癌细胞中用鸟嘌呤 (ED_miR-3144-3p(3_A < G)) 替换种子区域位置 3 的腺苷。然后,我们证明了Musashi RNA 结合蛋白 2 ( MSI2 ) 是 miR-3144-3p 的特定靶标,并且 MSI2 过表达是由于肝癌中典型 miR-3144-3p 过度依赖 ADAR1 的过度编辑所致。此外,目标预测分析和验证实验确定了溶质载体家族 38 个成员 4( SLC38A4 ) 作为 ED_miR-3144-3p(3_A < G) 的特定基因靶点。ADAR1 和 ED_miR-3144-3p(3_A < G) 的异位表达模拟增强的有丝分裂活动,ADAR1 抑制肝癌细胞中的 SLC38A4 表达。在自发性肝癌小鼠模型中,用小鼠特异性 ADAR1、MSI2-siRNA 或 SLC38A4 表达质粒进行治疗可抑制肿瘤发生和肿瘤生长。我们的研究结果表明,ADAR1 的异常调节通过过度编辑典型的 miR-3144-3p 来增强致癌 MSI2 效应,并且由此产生的 ED_miR-3144-3p(3_A < G) 同时抑制肿瘤抑制因子 SLC38A4 的表达,从而导致肝细胞癌发生。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
ADAR1-dependent miR-3144-3p editing simultaneously induces MSI2 expression and suppresses SLC38A4 expression in liver cancer
Aberrant adenosine-to-inosine (A-to-I) RNA editing, catalyzed by adenosine deaminase acting on double-stranded RNA (ADAR), has been implicated in various cancers, but the mechanisms by which microRNA (miRNA) editing contributes to cancer development are largely unknown. Our multistage hepatocellular carcinogenesis transcriptome data analyses, together with publicly available data, indicated that ADAR1 was the most profoundly dysregulated gene among RNA-editing enzyme family members in liver cancer. Targeted inactivation of ADAR1 inhibited the in vitro tumorigenesis of liver cancer cells. An integrative computational analyses of RNA-edited hotspots and the known editing frequency of miRNAs suggested that the miRNA miR-3144-3p was edited by ADAR1 during liver cancer progression. Specifically, ADAR1 promoted A-to-I editing of canonical miR-3144-3p to replace the adenosine at Position 3 in the seed region with a guanine (ED_miR-3144-3p(3_A < G)) in liver cancer cells. We then demonstrated that Musashi RNA-binding protein 2 (MSI2) was a specific target of miR-3144-3p and that MSI2 overexpression was due to excessive ADAR1-dependent over-editing of canonical miR-3144-3p in liver cancer. In addition, target prediction analyses and validation experiments identified solute carrier family 38 member 4 (SLC38A4) as a specific gene target of ED_miR-3144-3p(3_A < G). The ectopic expression of both ADAR1 and the ED_miR-3144-3p(3_A < G) mimic enhanced mitotic activities, and ADAR1 suppressed SLC38A4 expression in liver cancer cells. Treatments with mouse-specific ADAR1-, MSI2-siRNA-, or SLC38A4-expressing plasmids suppressed tumorigenesis and tumor growth in a mouse model of spontaneous liver cancer. Our findings suggest that the aberrant regulation of ADAR1 augments oncogenic MSI2 effects by excessively editing canonical miR-3144-3p and that the resultant ED_miR-3144-3p(3_A < G) simultaneously suppresses tumor suppressor SLC38A4 expression, contributing to hepatocellular carcinogenesis.
































 京公网安备 11010802027423号
京公网安备 11010802027423号