Journal of Molecular Modeling ( IF 2.1 ) Pub Date : 2023-01-03 , DOI: 10.1007/s00894-022-05435-x
Faheem Abbas 1 , Mohsen D Mohammadi 2 , Hitler Louis 3 , Ernest C Agwamba 3, 4
|
语境
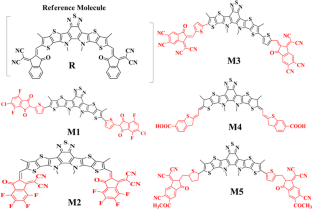
采用密度泛函理论 (DFT) 方法研究了苯并 [1,2,5] 噻二唑与 3,7-二甲基-3a,6,7,6,7, 7b-tetrahydro-5H-thieno[2',3':4,5]thieno[3,2-b]pyrrole 形成 3,9,12,13-tetramethyl-12,13-dihydro-[1,2, 5]噻二唑并[3,4-e]噻吩并[2″,3″:4,5]吡咯并[3.2-g]噻吩并[2′,3′:4,5]噻吩并[3,2-b]吲哚作为受体 (A),与作为 π-间隔基的噻吩桥接到供体部分 (D),它们是 2,3-二氢苯并 [b]噻吩-6-羧酸 (M4) 和功能化的 R、M1、M2、M3,和M5给出一个D-π-A-π-D。这是我们分子的反向组合:A-π-D-π-A 型发色团配置。还观察到调整多诺桥配置显着增加了电荷转移的便利性,因为能隙以 M4 中的 1.29 eV < M3 中的 1.59 eV < 1.67 eV < M2 中的 1.99 和 2.06 eV 的数量级减小。M3 (0.0031) 和 M5 (0.0031) 的重组能 (RE) 表示增加的顺序为 M3 > M5 > R > M2 > M4 > M1。HOMO-LUMO 表明,与设计的分子 M1-M5 相比,参考 R 在 0.990 eV 时反应性降低,而稳定性增加,其中 M1 在 0.970 eV 时最不稳定,而 M4 在 1.550 eV 时表现出最高稳定性. 设计分子的稳定性按M4:1.550 > M3:1.257 > M5:1.197 > M2:1.010 > M1:0.970 的顺序降低。所以,所有结果都表明缺电子核心是 M1-M5 化合物中有效的封端电子受体。作为通过降低 HOMO-LUMO 能级、重组能和结合能并提高这些设计分子的最大吸收和开路电压值来成功优化光电特性的理想配对。
方法
DFT 和 TDDFT 计算是用 Gaussian 16 程序进行的。使用具有 6-31 G (d,p) 基组的 CAM-B3LYP、WB97XD、B3LYP 和 MPW1PW91 泛函对建模化合物进行了全面优化。使用 PyMOlyze 软件绘制状态密度图。通过 Multiwfn 软件呈现其他分子特性,如过渡密度矩阵 (TDM) 和电子密度差图 (EDD)。
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号