当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational investigation of functionalized carbenes on dinitrogen activation
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-12-08 , DOI: 10.1002/jcc.27046 Justin K Kirkland 1 , Sophia K Johnson 1, 2 , Konstantinos D Vogiatzis 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-12-08 , DOI: 10.1002/jcc.27046 Justin K Kirkland 1 , Sophia K Johnson 1, 2 , Konstantinos D Vogiatzis 1
Affiliation
|
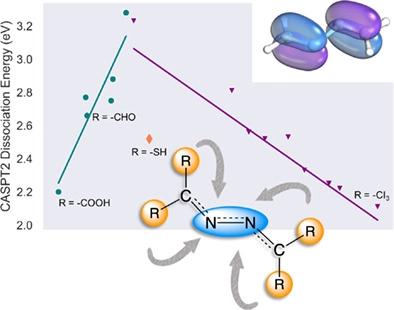
Activation of the dinitrogen triple bond is a crucial step in the overall fixation of atmospheric nitrogen into usable forms for industrial and biological applications. Current synthetic catalysts incorporate metal ions to facilitate the activation and cleavage of dinitrogen. The high price of metal-based catalysts and the challenge of catalyst recovery during industrial catalytic processes has led to increasing interest in metal-free catalysts. One step toward metal-free catalysis is the use of frustrated Lewis pairs (FLPs). In this study, we have examined 18 functionalized carbenes as FLPs to elucidate the influence of steric and electronic effects on the activation of dinitrogen. To test the effects of functionalization on dinitrogen activation, we have performed density functional theory (DFT), multireference, non and extended transition state-natural orbital for chemical valence (ETS-NOCV) calculations. Our results suggest that functional groups which introduce strong electron-withdrawing effects and/or engage in extended π/π* systems lead to the lowering of the dissociation energy of the dinitrogen bond, which further contributes to greater nitrogen activation. We conjecture that these effects are due to enhanced back-bonding capability of the p orbital of the carbene carbon atoms to the adjacent nitrogen atoms (increasing Lewis basicity of the carbene carbon atom) and enhanced stability of dissociated products. Our concluding remarks include opportunities to extend this activation study to explore the entire catalytic cycle with promising functionalized carbenes for experimental evaluation.
中文翻译:

功能化卡宾对双氮活化的计算研究
二氮三键的激活是将大气中的氮整体固定为工业和生物应用可用形式的关键步骤。目前的合成催化剂结合金属离子以促进二氮的活化和裂解。金属基催化剂的高价格和工业催化过程中催化剂回收的挑战导致人们对无金属催化剂的兴趣越来越大。迈向无金属催化的一步是使用受抑路易斯对 (FLP)。在这项研究中,我们检查了 18 种功能化卡宾作为 FLP,以阐明空间和电子效应对二氮活化的影响。为了测试功能化对二氮活化的影响,我们进行了密度泛函理论 (DFT)、多参考、用于化学价 (ETS-NOCV) 计算的非和扩展过渡态自然轨道。我们的结果表明,引入强吸电子效应和/或参与扩展的 π/π* 系统的官能团导致双氮键的解离能降低,这进一步有助于提高氮活化。我们推测这些效应是由于卡宾碳原子的 p 轨道与相邻氮原子的背键能力增强(增加卡宾碳原子的路易斯碱度)和解离产物的稳定性增强。我们的结束语包括扩展此活化研究的机会,以探索具有用于实验评估的有前途的功能化卡宾的整个催化循环。我们的结果表明,引入强吸电子效应和/或参与扩展的 π/π* 系统的官能团导致双氮键的解离能降低,这进一步有助于提高氮活化。我们推测这些效应是由于卡宾碳原子的 p 轨道与相邻氮原子的背键能力增强(增加卡宾碳原子的路易斯碱度)和解离产物的稳定性增强。我们的结束语包括扩展此活化研究的机会,以探索具有用于实验评估的有前途的功能化卡宾的整个催化循环。我们的结果表明,引入强吸电子效应和/或参与扩展的 π/π* 系统的官能团导致双氮键的解离能降低,这进一步有助于提高氮活化。我们推测这些效应是由于卡宾碳原子的 p 轨道与相邻氮原子的背键能力增强(增加卡宾碳原子的路易斯碱度)和解离产物的稳定性增强。我们的结束语包括扩展此活化研究的机会,以探索具有用于实验评估的有前途的功能化卡宾的整个催化循环。
更新日期:2022-12-08
中文翻译:
功能化卡宾对双氮活化的计算研究
二氮三键的激活是将大气中的氮整体固定为工业和生物应用可用形式的关键步骤。目前的合成催化剂结合金属离子以促进二氮的活化和裂解。金属基催化剂的高价格和工业催化过程中催化剂回收的挑战导致人们对无金属催化剂的兴趣越来越大。迈向无金属催化的一步是使用受抑路易斯对 (FLP)。在这项研究中,我们检查了 18 种功能化卡宾作为 FLP,以阐明空间和电子效应对二氮活化的影响。为了测试功能化对二氮活化的影响,我们进行了密度泛函理论 (DFT)、多参考、用于化学价 (ETS-NOCV) 计算的非和扩展过渡态自然轨道。我们的结果表明,引入强吸电子效应和/或参与扩展的 π/π* 系统的官能团导致双氮键的解离能降低,这进一步有助于提高氮活化。我们推测这些效应是由于卡宾碳原子的 p 轨道与相邻氮原子的背键能力增强(增加卡宾碳原子的路易斯碱度)和解离产物的稳定性增强。我们的结束语包括扩展此活化研究的机会,以探索具有用于实验评估的有前途的功能化卡宾的整个催化循环。我们的结果表明,引入强吸电子效应和/或参与扩展的 π/π* 系统的官能团导致双氮键的解离能降低,这进一步有助于提高氮活化。我们推测这些效应是由于卡宾碳原子的 p 轨道与相邻氮原子的背键能力增强(增加卡宾碳原子的路易斯碱度)和解离产物的稳定性增强。我们的结束语包括扩展此活化研究的机会,以探索具有用于实验评估的有前途的功能化卡宾的整个催化循环。我们的结果表明,引入强吸电子效应和/或参与扩展的 π/π* 系统的官能团导致双氮键的解离能降低,这进一步有助于提高氮活化。我们推测这些效应是由于卡宾碳原子的 p 轨道与相邻氮原子的背键能力增强(增加卡宾碳原子的路易斯碱度)和解离产物的稳定性增强。我们的结束语包括扩展此活化研究的机会,以探索具有用于实验评估的有前途的功能化卡宾的整个催化循环。






























 京公网安备 11010802027423号
京公网安备 11010802027423号