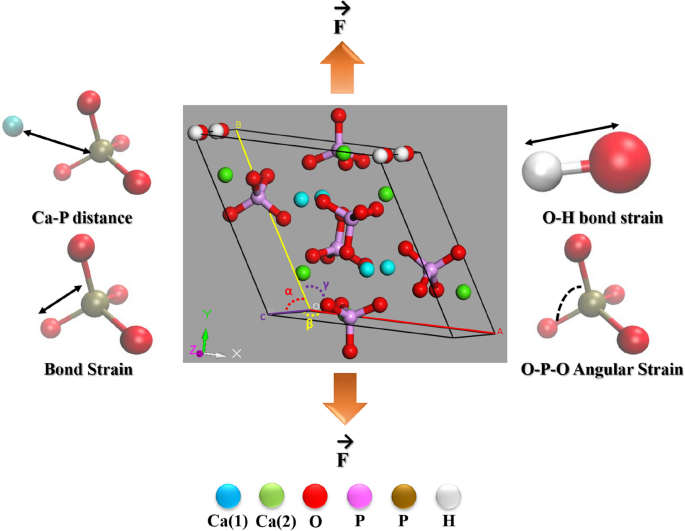
羟基磷灰石(HA,Ca 10 PO 4 (OH) 2 )因其与天然骨矿物质的相似性而成为生物材料科学实验领域中广泛探索的材料。具体而言,在生物陶瓷界,掺杂多价阳离子(例如Mg 2+ 、Fe 2+ 、Sr 2+等)的HA在过去几十年中得到了广泛的研究。实验研究很大程度上确定了掺杂剂含量对机械和生物相容性的关键作用。掺杂 HA 机械响应的大量实验测量都是基于多晶材料的压缩或压痕测试。此类测量,更重要的是,单晶(掺杂)HA 机械性能的计算预测很少。在此前提下,本研究旨在建立不同 Fe 含量(10、20、30 和 40 mol%)的 Fe 2+掺杂 HA 的原子模型,并通过分子动力学(MD)探索其单轴拉伸响应。 ) 模拟。在平衡的晶胞结构中,发现 Ca(1) 位点在能量上有利于 Fe 2+取代。 Fe 2+离子的局部分布显着影响官能团周围的原子部分电荷分布和化学对称性,并且在MD分析的红外光谱中发现了这样的特征。由于晶体结构的对称性变化,Fe 掺杂 HA 中的红外谱带强度显着降低,同时出现谱带分裂。这项工作的另一个重要目标是通过计算预测单晶形式的掺杂 HA 的机械响应。 一个有趣的观察是,未掺杂 HA 的弹性各向异性并未因 Fe 掺杂而受到影响。在具有 Fe 2+掺杂剂含量的掺杂 HA 中,拉伸强度 (TS) 系统性降低,并且 TS 随温度的降低可归因于高温下原子热搅动的增加。拉伸响应的物理原理根据共价/离子键框架的应变相关变化(Ca-P距离、P-O键应变、O-P-O角应变、O-H键距离)合理化。此外,通过计算O-H和P-O键振动能的变化,对共价键网络的动态变化进行了能量分析。总而言之,当前的工作建立了我们对铁掺杂 HA 单晶的结构稳定性和拉伸响应所涉及的原子现象的基础理解。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
In silico study on probing atomistic insights into structural stability and tensile properties of Fe-doped hydroxyapatite single crystals
Hydroxyapatite (HA, Ca10PO4(OH)2) is a widely explored material in the experimental domain of biomaterials science, because of its resemblance with natural bone minerals. Specifically, in the bioceramic community, HA doped with multivalent cations (e.g., Mg2+, Fe2+, Sr2+, etc.) has been extensively investigated in the last few decades. Experimental research largely established the critical role of dopant content on mechanical and biocompatibility properties. The plethora of experimental measurements of mechanical response on doped HA is based on compression or indentation testing of polycrystalline materials. Such measurements, and more importantly the computational predictions of mechanical properties of single crystalline (doped) HA are scarce. On that premise, the present study aims to build atomistic models of Fe2+-doped HA with varying Fe content (10, 20, 30, and 40 mol%) and to explore their uniaxial tensile response, by means of molecular dynamics (MD) simulation. In the equilibrated unit cell structures, Ca(1) sites were found to be energetically favourable for Fe2+ substitution. The local distribution of Fe2+ ions significantly affects the atomic partial charge distribution and chemical symmetry surrounding the functional groups, and such signatures are found in the MD analyzed IR spectra. The significant decrease in the intensity of the IR bands found in the Fe-doped HA together with band splitting, because of the symmetry changes in the crystal structure. Another important objective of this work is to computationally predict the mechanical response of doped HA in their single crystal format. An interesting observation is that the elastic anisotropy of undoped HA was not compromised with Fe-doping. Tensile strength (TS) is systematically reduced in doped HA with Fe2+ dopant content and a decrease in TS with temperature can be attributed to the increased thermal agitation of atoms at elevated temperatures. The physics of the tensile response was rationalized in terms of the strain dependent changes in covalent/ionic bond framework (Ca–P distance, P–O bond strain, O–P–O angular strain, O–H bond distance). Further, the dynamic changes in covalent bond network were energetically analyzed by calculating the changes in O–H and P–O bond vibrational energy. Summarizing, the current work establishes our foundational understanding of the atomistic phenomena involved in the structural stability and tensile response of Fe-doped HA single crystals.
































 京公网安备 11010802027423号
京公网安备 11010802027423号