Minerals Engineering ( IF 4.9 ) Pub Date : 2022-11-21 , DOI: 10.1016/j.mineng.2022.107935 Reece R. Waltrovitz , Gujie Qian , Frank de Groot , Jamie S. Quinton , Sarah L. Harmer
|
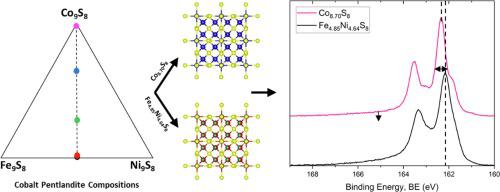
X-ray photoemission spectroscopy (XPS) was used to investigate the electronic structure and oxidation state of transition metals in synthetic cobalt pentlandites (Fe,Co,Ni)9S8 with measured stoichiometries of Fe4.85Ni4.64S8, Co0.13Fe4.68Ni4.71S8, Co2.73Fe3.18Ni3.16S8, Co5.80Fe1.63Ni1.59S8 and Co8.70S8. The addition of cobalt was found to decrease the bond length and hence decrease the unit cell dimensions of the cobalt pentlandite crystal structure by up to 0.18 Å. High resolution XPS S 2p spectra show increases in the binding energies of bulk 5-coordinated (162.1 eV) and surface 3-coordinated (160.9 eV) sulfur (≈0.2 eV) with increasing Co concentration and decreasing bond lengths. As the Co concentration increases, variation in metal site occupation decreases, resulting in smaller S 2p FWHMs due to a dominant single Co-S state rather than mixed Fe-S, Ni-S and Co-S states with similar binding energies. The S 2p high binding energy tail, previously identified as a S 3p- Fe 3d ligand to metal transfer in Fe chalcogenides, shows a marked decrease in intensity as the concentration of Co increases, that is attributed to a decreased probability of ligand-to-metal charge transfer as the eg orbitals are filled. The transition metal XPS 2p spectra were modelled using CTM4XAS to investigate metal site occupation and ligand-to-metal charge transfer. Fe, Co, and Ni were all best simulated using a tetrahedral symmetry and 2+ oxidation state, their 2p3/2 and 2p1/2 peaks occurred at 706.9 and 719.9 eV, 778.2 and 793.1 eV, and 852.8 and 870.0 eV, respectively. A negative charge transfer energy confirms the high binding energy tail results from S3p-Fe3d ligand to metal charge transfer. This increased understanding of the pentlandite electronic structure will provide a basis for the refinement of mineral processing techniques and allow for a reduced environmental impact from limited efficiency.
中文翻译:
Co:Fe:Ni比值对钴镍黄铁矿电子结构和表面形态的影响
X 射线光电子能谱 (XPS) 用于研究合成钴镍黄铁矿 (Fe,Co,Ni) 9 S 8中过渡金属的电子结构和氧化态,并测量了 Fe 4.85 Ni 4.64 S 8、Co 0.13 Fe 4.68的化学计量Ni 4.71 S 8、 Co 2.73 Fe 3.18 Ni 3.16 S 8、 Co 5.80 Fe 1.63 Ni 1.59 S 8和 Co 8.70 S 8。发现添加钴会降低键长,因此会降低钴镍黄铁矿晶体结构的晶胞尺寸高达 0.18 Å。高分辨率 XPS S 2 p光谱显示,随着 Co 浓度的增加和键长的减少,本体 5 配位 (162.1 eV) 和表面 3 配位 (160.9 eV) 硫 (≈0.2 eV) 的结合能增加。随着 Co 浓度的增加,金属位点占据的变化减少,导致更小的 S 2 p FWHM,这是由于主要的单一 Co-S 状态而不是具有相似结合能的混合 Fe-S、Ni-S 和 Co-S 状态。S 2 p高结合能尾部,之前被确定为 S 3 p - Fe 3 dFe 硫属元素化物中配体到金属的转移表明,随着 Co 浓度的增加,强度显着降低,这归因于随着 e g轨道被填充,配体到金属电荷转移的可能性降低。使用 CTM4XAS 对过渡金属 XPS 2 p光谱进行建模,以研究金属位点占据和配体到金属的电荷转移。Fe、Co 和 Ni 都使用四面体对称性和 2+ 氧化态进行了最佳模拟,它们的 2p 3/2和 2p 1/2峰分别出现在 706.9 和 719.9 eV、778.2 和 793.1 eV 以及 852.8 和 870.0 eV 处. 负电荷转移能证实了 S3 p -Fe3 d的高结合能尾部结果配体到金属的电荷转移。这种对镍黄铁矿电子结构的深入了解将为改进矿物加工技术提供基础,并减少因效率有限而对环境造成的影响。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号