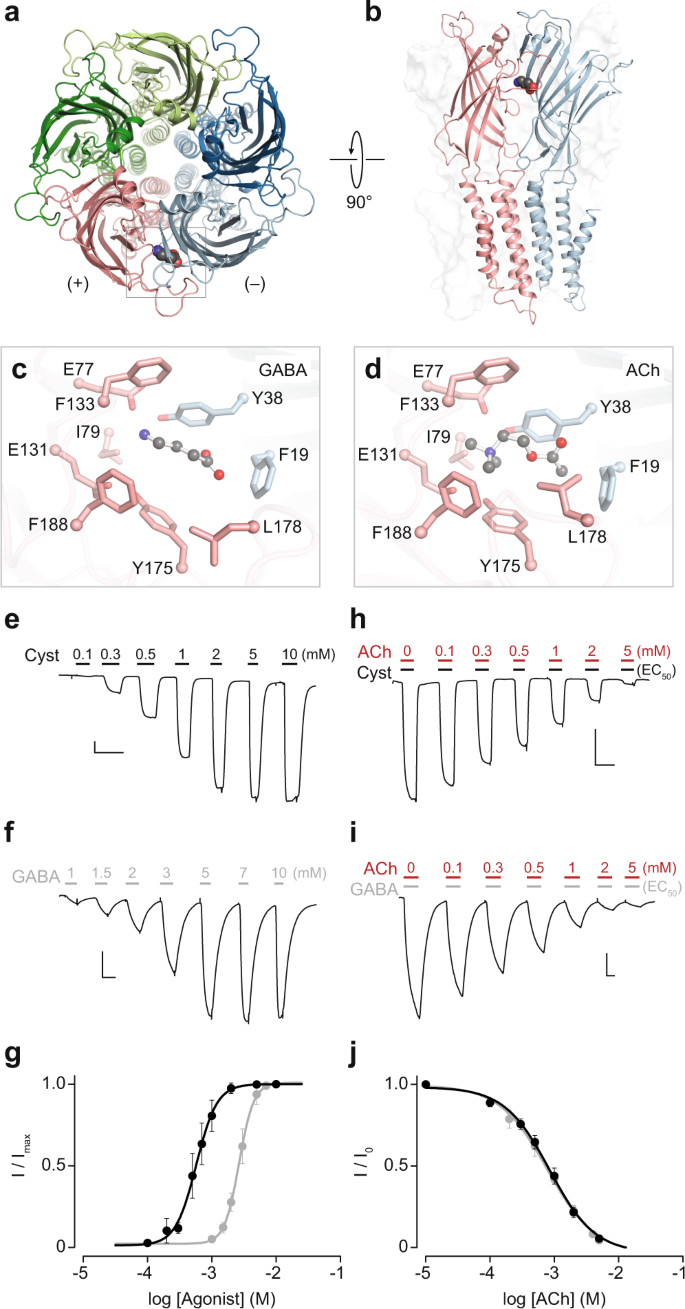
ELIC 是一种原核同源五聚体配体门控离子通道,与脊椎动物烟碱乙酰胆碱受体同源。乙酰胆碱与 ELIC 结合但未能激活它,尽管带来了指示激活的构象变化。相反,乙酰胆碱竞争性地抑制激动剂激活的 ELIC 电流。是什么让乙酰胆碱成为乙酰胆碱受体环境中的激动剂,以及 ELIC 环境中的拮抗剂,尚不清楚。在这里,我们使用可用的结构和统计耦合分析来识别 ELIC 激动剂结合位点中有助于激动的残基。用这些 ELIC 残基取代它们的乙酰胆碱受体对应物不会将乙酰胆碱转化为 ELIC 激动剂,但在某些情况下会降低 ELIC 对乙酰胆碱拮抗作用的敏感性。乙酰胆碱拮抗作用可以通过组合两个似乎共同敲除乙酰胆碱结合的取代来消除。因此,使 ELIC 激动剂结合位点更像乙酰胆碱受体,反常地降低了对乙酰胆碱的表观亲和力,表明在一种情况下对激动剂结合重要的残基在另一种情况下可能是有害的。这些发现强化了这样一种观念,即尽管激动作用起源于激动剂结合位点内的局部相互作用,但它是一种全局特性,具有来自远处残基的神秘贡献。最后,我们的结果强调了一种被低估的对抗机制,其中 反常地降低了对乙酰胆碱的表观亲和力,表明在一种情况下对激动剂结合很重要的残基在另一种情况下可能是有害的。这些发现强化了这样一种观念,即尽管激动作用起源于激动剂结合位点内的局部相互作用,但它是一种全局特性,具有来自远处残基的神秘贡献。最后,我们的结果强调了一种被低估的对抗机制,其中 反常地降低了对乙酰胆碱的表观亲和力,表明在一种情况下对激动剂结合很重要的残基在另一种情况下可能是有害的。这些发现强化了这样一种观念,即尽管激动作用起源于激动剂结合位点内的局部相互作用,但它是一种全局特性,具有来自远处残基的神秘贡献。最后,我们的结果强调了一种被低估的对抗机制,其中具有可观亲和力但功效可忽略不计的激动剂作为竞争性拮抗剂存在。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Origin of acetylcholine antagonism in ELIC, a bacterial pentameric ligand-gated ion channel
ELIC is a prokaryotic homopentameric ligand-gated ion channel that is homologous to vertebrate nicotinic acetylcholine receptors. Acetylcholine binds to ELIC but fails to activate it, despite bringing about conformational changes indicative of activation. Instead, acetylcholine competitively inhibits agonist-activated ELIC currents. What makes acetylcholine an agonist in an acetylcholine receptor context, and an antagonist in an ELIC context, is not known. Here we use available structures and statistical coupling analysis to identify residues in the ELIC agonist-binding site that contribute to agonism. Substitution of these ELIC residues for their acetylcholine receptor counterparts does not convert acetylcholine into an ELIC agonist, but in some cases reduces the sensitivity of ELIC to acetylcholine antagonism. Acetylcholine antagonism can be abolished by combining two substitutions that together appear to knock out acetylcholine binding. Thus, making the ELIC agonist-binding site more acetylcholine receptor-like, paradoxically reduces the apparent affinity for acetylcholine, demonstrating that residues important for agonist binding in one context can be deleterious in another. These findings reinforce the notion that although agonism originates from local interactions within the agonist-binding site, it is a global property with cryptic contributions from distant residues. Finally, our results highlight an underappreciated mechanism of antagonism, where agonists with appreciable affinity, but negligible efficacy, present as competitive antagonists.






























 京公网安备 11010802027423号
京公网安备 11010802027423号