当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Influence of Molecular Configurations on the Desulfonylation Reactions on Metal Surfaces
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2022-11-16 , DOI: 10.1021/jacs.2c08736 Junbo Wang 1, 2 , Kaifeng Niu 1, 3 , Chaojie Xu 1 , Huaming Zhu 2 , Honghe Ding 4 , Dong Han 4 , Yuanjing Zheng 1 , Jiahao Xi 1 , Sifan You 1 , Chuan Deng 2 , Haiping Lin 2 , Johanna Rosen 3 , Junfa Zhu 4 , Jonas Björk 3 , Qing Li 2 , Lifeng Chi 1, 5
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2022-11-16 , DOI: 10.1021/jacs.2c08736 Junbo Wang 1, 2 , Kaifeng Niu 1, 3 , Chaojie Xu 1 , Huaming Zhu 2 , Honghe Ding 4 , Dong Han 4 , Yuanjing Zheng 1 , Jiahao Xi 1 , Sifan You 1 , Chuan Deng 2 , Haiping Lin 2 , Johanna Rosen 3 , Junfa Zhu 4 , Jonas Björk 3 , Qing Li 2 , Lifeng Chi 1, 5
Affiliation
|
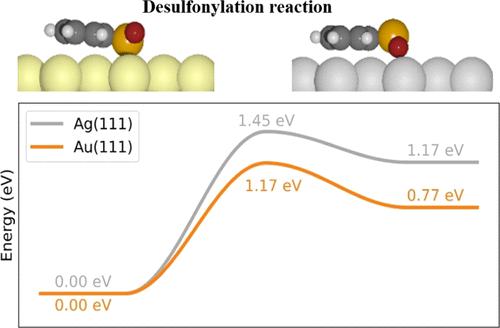
On-surface synthesis is a powerful methodology for the fabrication of low-dimensional functional materials. The precursor molecules usually anchor on different metal surfaces via similar configurations. The activation energies are therefore solely determined by the chemical activity of the respective metal surfaces. Here, we studied the influence of the detailed adsorption configuration on the activation energy on different metal surfaces. We systematically studied the desulfonylation homocoupling for a molecular precursor on Au(111) and Ag(111) and found that the activation energy is lower on inert Au(111) than on Ag(111). Combining scanning tunneling microscopy observations, synchrotron radiation photoemission spectroscopy measurements, and density functional theory calculations, we elucidate that the phenomenon arises from different molecule–substrate interactions. The molecular precursors anchor on Au(111) via Au–S interactions, which lead to weakening of the phenyl–S bonds. On the other hand, the molecular precursors anchor on Ag(111) via Ag–O interactions, resulting in the lifting of the S atoms. As a consequence, the activation barrier of the desulfonylation reactions is higher on Ag(111), although silver is generally more chemically active than gold. Our study not only reports a new type of on-surface chemical reaction but also clarifies the influence of detailed adsorption configurations on specific on-surface chemical reactions.
中文翻译:

分子构型对金属表面脱磺酰化反应的影响
表面合成是制造低维功能材料的有效方法。前体分子通常通过以下方式锚定在不同的金属表面上类似的配置。因此,活化能仅由相应金属表面的化学活性决定。在这里,我们研究了详细的吸附构型对不同金属表面活化能的影响。我们系统地研究了分子前体在 Au(111) 和 Ag(111) 上的脱磺酰化自偶联,发现惰性 Au(111) 上的活化能低于 Ag(111) 上的活化能。结合扫描隧道显微镜观察、同步辐射光电子能谱测量和密度泛函理论计算,我们阐明了这种现象是由不同的分子-底物相互作用引起的。分子前体通过 Au(111) 锚定Au-S相互作用,导致苯基-S键弱化。另一方面,分子前体通过Ag-O 相互作用锚定在 Ag(111) 上,导致 S 原子的提升。因此,Ag(111) 上脱磺酰化反应的活化能垒更高,尽管银通常比金具有更高的化学活性。我们的研究不仅报告了一种新型的表面化学反应,而且阐明了详细的吸附配置对特定表面化学反应的影响。
更新日期:2022-11-16
中文翻译:
分子构型对金属表面脱磺酰化反应的影响
表面合成是制造低维功能材料的有效方法。前体分子通常通过以下方式锚定在不同的金属表面上类似的配置。因此,活化能仅由相应金属表面的化学活性决定。在这里,我们研究了详细的吸附构型对不同金属表面活化能的影响。我们系统地研究了分子前体在 Au(111) 和 Ag(111) 上的脱磺酰化自偶联,发现惰性 Au(111) 上的活化能低于 Ag(111) 上的活化能。结合扫描隧道显微镜观察、同步辐射光电子能谱测量和密度泛函理论计算,我们阐明了这种现象是由不同的分子-底物相互作用引起的。分子前体通过 Au(111) 锚定Au-S相互作用,导致苯基-S键弱化。另一方面,分子前体通过Ag-O 相互作用锚定在 Ag(111) 上,导致 S 原子的提升。因此,Ag(111) 上脱磺酰化反应的活化能垒更高,尽管银通常比金具有更高的化学活性。我们的研究不仅报告了一种新型的表面化学反应,而且阐明了详细的吸附配置对特定表面化学反应的影响。






























 京公网安备 11010802027423号
京公网安备 11010802027423号