Structural Chemistry ( IF 2.1 ) Pub Date : 2022-10-17 , DOI: 10.1007/s11224-022-02033-8 Rajan Jeevana 1 , Abu Pilakkaveettil Kavitha 2 , Thoppilan G Abi 3 , Pookkottu K Sajith 2 , Jibin K Varughese 3 , Kuttamath Kunniyur Aravindakshan 4
|
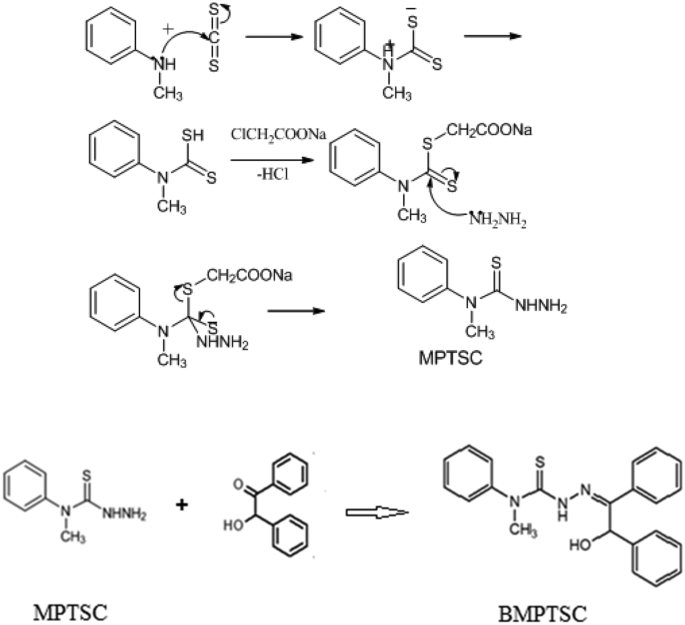
The global spread of the COVID-19 pandemic caused by the etiological agent, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), triggered researchers to identify and develop novel antiviral therapeutics. Herein, we report a new molecule 2-hydroxy-1,2-diphenylethanone N(4)-methyl-N(4)-phenyl thiosemicarbazone (BMPTSC), as a potential inhibitor of SARS-CoV-2. BMPTSC was synthesized, characterized by IR and NMR studies, and the structural parameters were analyzed computationally by B3LYP/cc-pVDZ method. Molecular docking studies were performed to get insights into the energetics and compatibility of BMPTSC against various SARS-CoV-2 drug targets. The best docking poses of target protein-BMPTSC complex structures were further subjected to molecular dynamics (MD) simulations. Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) calculations on the binding of BMPTSC with the target proteins viz. spike glycoprotein and ACE-2 protein showed energy values of −179.87 and −145.61 kJ/mol, respectively. Moreover, BMPTSC obeys Lipinski’s rule, and further in silico assessment of oral bioavailability, bioactivity scores, ADME, drug-likeness, and medicinal chemistry friendliness suggests that this molecule is a promising candidate for the COVID-19 drug discovery process.
中文翻译:
针对 COVID-19 大流行:2-羟基-1, 2-二苯基乙酮 N(4)-甲基-N(4)-缩氨基苯硫脲作为 SARS-CoV-2 潜在抑制剂的计算机评估
由病原体严重急性呼吸综合征冠状病毒-2 (SARS-CoV-2) 引起的 COVID-19 大流行在全球蔓延,促使研究人员识别和开发新型抗病毒疗法。在此,我们报告了一种新分子 2-羟基-1,2-二苯基乙酮 N(4)-甲基-N(4)-苯基缩氨基硫脲 (BMPTSC),作为 SARS-CoV-2 的潜在抑制剂。合成了BMPTSC,通过IR和NMR研究进行了表征,并通过B3LYP/cc-pVDZ方法对结构参数进行了计算分析。进行分子对接研究是为了深入了解 BMPTSC 针对各种 SARS-CoV-2 药物靶点的能量学和相容性。目标蛋白-BMPTSC复合物结构的最佳对接姿势进一步进行分子动力学(MD)模拟。 BMPTSC 与目标蛋白结合的分子力学泊松-玻尔兹曼表面积 (MM-PBSA) 计算。刺突糖蛋白和ACE-2蛋白的能量值分别为-179.87和-145.61 kJ/mol。此外,BMPTSC 遵循 Lipinski 规则,并且进一步对口服生物利用度、生物活性评分、ADME、药物相似性和药物化学友好性进行计算机评估,表明该分子是 COVID-19 药物发现过程的有希望的候选者。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号