当前位置:
X-MOL 学术
›
J. Mol. Liq.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
咪唑基离子液体在 Fe(1 0 0) 表面的吸附以抑制腐蚀:物理吸附还是化学吸附?
Journal of Molecular Liquids ( IF 5.3 ) Pub Date : 2022-09-30 , DOI: 10.1016/j.molliq.2022.120489
Meng-Fu Chen , Yingqian Chen , Zhen Jia Lim , Ming Wah Wong
|
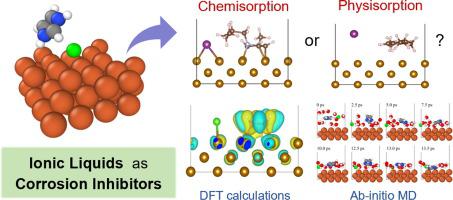
使用基于第一性原理的密度泛函理论 (DFT) 计算和从头开始分子动力学 (AIMD) 研究了咪唑基离子液体 (ILs) 作为铁的缓蚀剂的使用,以了解它们的吸附结构、稳定性和机理。共筛选出 36 种离子液体 (ILs),具有不同阴离子类型(Cl−、Br−、I−、N(CN)2−、BF4− 和 PF6−)和烷基链长度(Cn、n = 0、1、2、4 和 6)。我们的计算表明,ILs 在金属表面表现出很强的吸附性,在各种不同的标准下被归类为化学吸附。AIMD 模拟包括显式溶剂化效应,验证了在真空下计算的这些吸附构型代表了水性环境中的吸附过程。此外,我们表明这种化学吸附相互作用的基础是通过从铁板到咪唑鎓环的净电子捐赠。采用分散校正 DFT 方法 (DFT-D3),我们证明了阳离子的烷基链长度对吸附能有很大影响。最后,我们表明,与经典的分子动力学计算相比,DFT 计算对吸附过程的建模要准确得多。这项计算研究的结果与过去的实验结果一致,并表明 DFT 计算可用于深入了解参与腐蚀抑制的绿色和可持续 IL 的吸附过程的性质。
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号