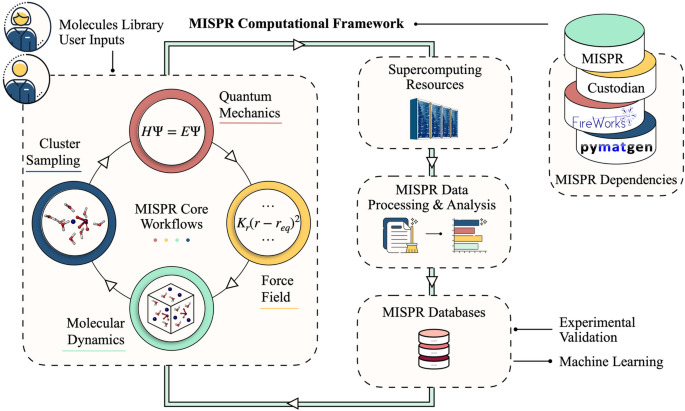
计算工具通过增强我们理解原子结构和功能特性之间联系的能力,为研究和设计最佳材料提供了独特的机会。然而,由于需要集成各种计算化学软件(不一定彼此兼容)、生成数据的异构性以及需要探索巨大的化学和参数空间,设计具有定制功能的材料是复杂的。后者对于避免基于分散数据点的模型中的偏差并导出只能通过系统数据集访问的统计趋势尤其重要。在这里,我们引入了一个强大的高通量多尺度计算基础设施,称为 MISPR(结构-性能关系材料信息学),它将经典分子动力学(MD)模拟与密度泛函理论(DFT)无缝集成。通过实现高性能数据分析以及不同方法和规模之间的耦合,MISPR 解决了自动化工作流程管理和数据来源记录需求所带来的关键挑战。 MISPR 的主要功能包括自动 DFT 和 MD 模拟、错误处理、分子和系综属性的推导,以及创建输出数据库来组织各个计算的结果以实现可重复性和透明度。在这项工作中,我们描述了在 MISPR 中实现的全自动 DFT 工作流程,以计算各种属性,例如核磁共振化学位移、结合能、键解离能和氧化还原电位,并支持电子转移和质子耦合电子转移等多种方法反应。 该基础设施还通过提供计算液体溶液中各种结构和动力学特性的 MD 工作流程来表征大规模系综特性。 MISPR 采用材料信息学方法来促进现象结构-性能关系的理解和预测,这对于为众多科学应用和工程技术设计新颖的最佳材料至关重要。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
MISPR: an open-source package for high-throughput multiscale molecular simulations
Computational tools provide a unique opportunity to study and design optimal materials by enhancing our ability to comprehend the connections between their atomistic structure and functional properties. However, designing materials with tailored functionalities is complicated due to the necessity to integrate various computational-chemistry software (not necessarily compatible with one another), the heterogeneous nature of the generated data, and the need to explore vast chemical and parameter spaces. The latter is especially important to avoid bias in scattered data points-based models and derive statistical trends only accessible by systematic datasets. Here, we introduce a robust high-throughput multi-scale computational infrastructure coined MISPR (Materials Informatics for Structure–Property Relationships) that seamlessly integrates classical molecular dynamics (MD) simulations with density functional theory (DFT). By enabling high-performance data analytics and coupling between different methods and scales, MISPR addresses critical challenges arising from the needs of automated workflow management and data provenance recording. The major features of MISPR include automated DFT and MD simulations, error handling, derivation of molecular and ensemble properties, and creation of output databases that organize results from individual calculations to enable reproducibility and transparency. In this work, we describe fully automated DFT workflows implemented in MISPR to compute various properties such as nuclear magnetic resonance chemical shift, binding energy, bond dissociation energy, and redox potential with support for multiple methods such as electron transfer and proton-coupled electron transfer reactions. The infrastructure also enables the characterization of large-scale ensemble properties by providing MD workflows that calculate a wide range of structural and dynamical properties in liquid solutions. MISPR employs the methodologies of materials informatics to facilitate understanding and prediction of phenomenological structure–property relationships, which are crucial to designing novel optimal materials for numerous scientific applications and engineering technologies.
































 京公网安备 11010802027423号
京公网安备 11010802027423号